Aşağıdaki mikrodelesyon sendromlarından hangisine lizensefali eşlik eder?
A) Williams sendromu
B) Prader Willi sendromu
C) DiGeorge sendromu
D) Miller-Dieker sendromu*****
E) Alagille sendromu
Lizensefali girusların yokluğu ile karakterizedir. Miller-Dicker sendromunun en karakteristik bulgusu lizensefalidir. Miller-Dicker sendromunda lizensefaliden başka mikrosefali, pakigiri, nöbetler, hipogenitaliya ve mental retardasyon görülebilir.
Miller–Dieker Sendromu (Lissensefali Tip 1)
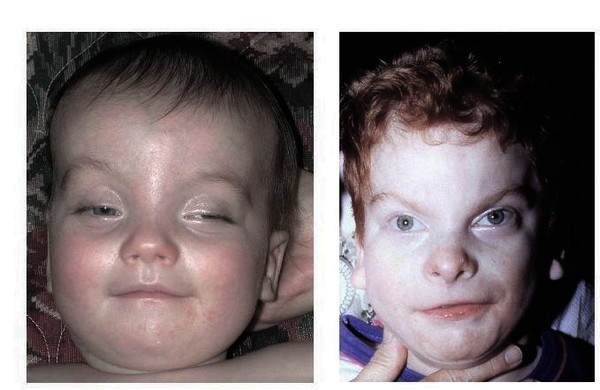
Miller–Dieker sendromu, ağır bir klasik (tip 1) lissensefali tablosudur. Temel özellik; Beynin yüzeyinin düz olması (agiri–pakigiri) + tipik yüz görünümü gösterir.
Genetik Temel
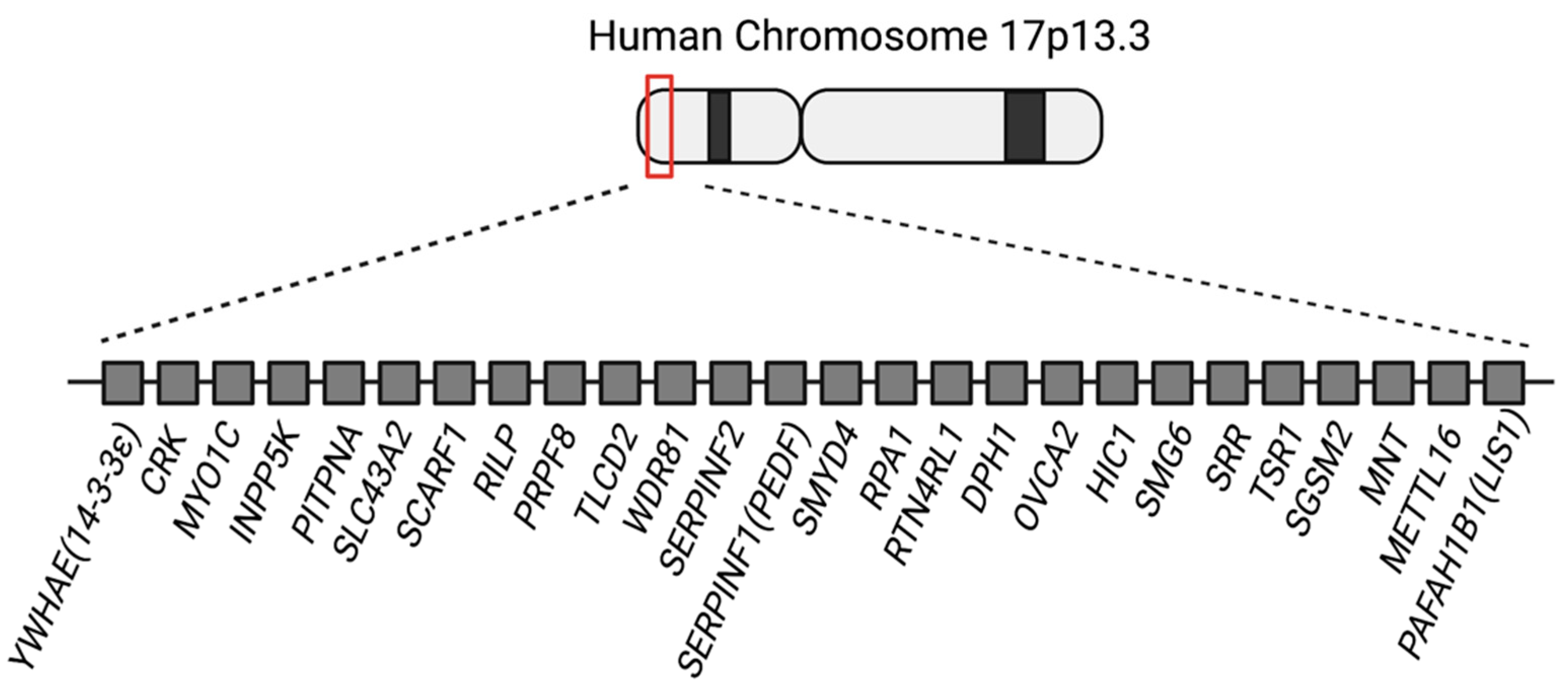
Kromozom, 17p13.3 delesyonudur. Bu noktada rol alan kritik gen; LIS1 (PAFAH1B1) dir.
Nöronal migrasyon bozukluğuna bağlı Korteks düzgün kıvrım yapamaz.
Patogenez; Normalde 12–24. gebelik haftasında nöronlar ventriküler zon’dan kortekse göç eder. Miller–Dieker’de ise Nöronal migrasyon bozulur, 6 katlı korteks oluşamaz ve Beyin yüzeyi düz kalır
Klinik Bulgular
| Bulgular | Açıklama |
|---|---|
| Şiddetli mental retardasyon | Ağır |
| Epilepsi | İnfantil spazm sık |
| Hipotoni | Erken dönemde |
| Beslenme güçlüğü | Sık |
| Mikrosefali | Olabilir |
Tipik Yüz Görünümü; Yüksek alın, Bitemporal daralma, Küçük çene (mikrognati), Yukarı dönük burun ve İnce üst dudak şeklinde tanımlanır.
Görüntüleme Bulguları
MRI de Agiri (gyrus yokluğu), Pakigiri, Kortikal kalınlaşma ve Düz beyin yüzeyidir.
Ayırıcı Tanı
| Hastalık | Fark |
|---|---|
| İzole lissensefali | Yüz dismorfisi yok |
| Walker-Warburg | Göz anomalisi + kas distrofisi |
| Zellweger | Peroksizomal hastalık |
TUS İpuçları
17p delesyonu → Miller–Dieker
Düz beyin (smooth brain) → Lissensefali
İnfantil spazm → Sık eşlik eder
Tipik yüz + lissensefali → Sendromik form
Klinik Mini Vaka
3 aylık bebekte İnfantil spazm, Şiddetli hipotoni, MRI de agiri ve Dismorfik yüz var ise Tanı: Miller–Dieker sendromu