Son 3 yıldır generalize kore izlenen 26 yaş kadın hastanın dedesinin 52 yaşında başlayan hareket bozukluğu, psikoz ve demans nedeniyle 78 yaşında vefat ettiği, şimdi 52 yaşında olan babasının da 11 yıldır giderek artan istemsiz hareketleri ve unutkanlığı olduğu öğreniliyor.
Bu hastadaki en olası tanı için hangisi doğru değildir?
A) O.D geçişli bir hastalığa sahiptir.
B) Antisipasyon göstermektedir.
C) Babadan kalıtıldıkça daha erken yaşta başlamaktadır.
D) Elektronöromiyografi tanı koymada yararsızdır.
E) Trinükleotid tekrar (TNT) hastalığı olup CTG tekrarları dikkati çekmektedir.***************
Huntıngton koresi (poliglutamin hastalığı)
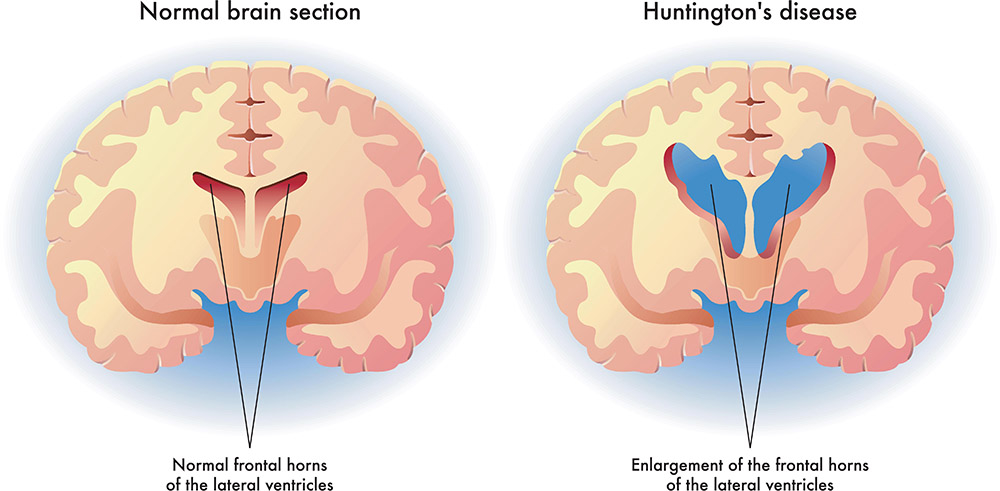
Sitriatumun (nükleus Caudatus ve putamen) dejenerasyonu ile giden 30-55 yaş, K=E, O.D, trinükleotid tekrar (C-A-G->40 tekrar ) hastalığıdır.
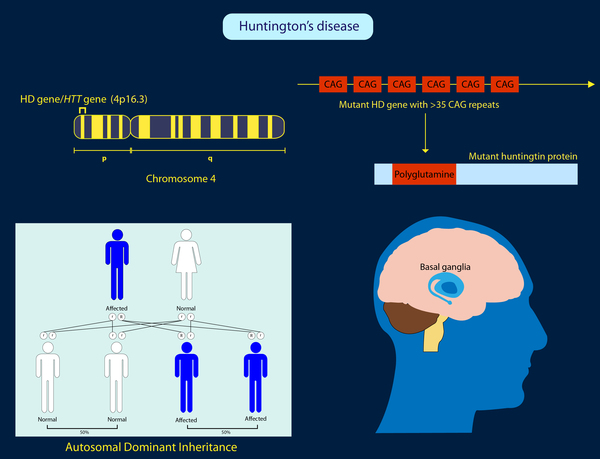
HH poliglutamin trinükleotid tekrar hastalığıdır. HH, huntington proteinini kodlayan 4p16.3 yerleşimli gendeki CAG trinükleotid tekrarları nedeniyle gelişir. Trinükleotid tekrar artışı ile giden hastalıklar arasında glutamini kodlayan CAG tekrar artışı ile karakterizepoliglutamin hastalıklarının prototipidir.
Burada yapısı etkilenen protein “huntingtin” dir, genetik bozukluk protein işlevinibozmaktan çok ona patolojik bir nitelik kazandırarak patolojiye yol açar. Bu gene ait CAG tekrar sayısı 37-39 üzerine çıkınca hastalıkoluşur ve sayı ne kadar yükselirse klinik o kadar erken başlangıçlı, hızlı ve şiddetli olur.
Mayoz bölünme sırasında tekrar sayısının ekspansiyonu böylece klinikte kuşaklar boyunca giderek erken yaşa çekilen ve şiddetlenen bir klinik görünüme yol açar (antisipasyon).
Premutasyon değerleri olan 35-37 arasında hastalık manifest olmaz ama bir sonraki kuşakta tekrar sayısı artarak kendini gösterir.
Patolojik sınırdaki mutasyonun geçirgenliği tamdır (%100), bu nedenle anne veya babası hasta olan bireyler %50 risk altındadır.
Bu durumdaki sağlam bireylerde genetik test (prediktif test) kesinlikle 18 yaş üzerinde ve mutlaka iyi bir genetik danışmanlık ve psikiyatrik değerlendirme sonrasında yapılmalıdır.
Hastalık genelde 3-4. dekadlarda başlar ve 15-20 yıl kadar sürer, ancak başlangıç temelde CAG tekrar sayısına bağlı olarak 5 yaşa kadar inebileceği gibi 70 yaş sonrasına da kalabilir.
Striatum hasarı (Caudat ve putamen) → kore-atetoid hareketler
Ach ↓, GABA ↓, Glutamat ↑, Dopamin ↑ sonucu NMDA toksisitesiyle nucleus Caudatus ve putamende Ach ve GABA düşer Caudat atrofi oluşur.
Depresyon (EN SIK), anksiyete, obsesif kompulsif semptomlar, psikoz ve artmış intihar girişimleri hastalıkta sıktır ama ölüm en sık enfeksiyon nedenli.
Vücudun tüm bölgelerinde istemsiz sıçrayıcı hareketler, ekstremitelerde kıvranır tarzda hareketler ve progresif bunama (demans) ile karakterizedir.
Hastalar ortalama 15 yılda ölür.
Kişilik değişiklikleri (Glutamat ↑) frontal kortekste atrofi ilişkili olabilir.
Genotip-fenotip korelasyonu çok kuvvetlidir, daha yüksek tekrar sayıları daha erken başlangıçlı hastalıkla sonuçlanır.
Beyin küçük, nükleus kaudatusta ve putamende belirgin atrofi dikkati çeker.
Korenin tedavisi için tetrabenazin (Dopamin antagonistleri)
