
Otuziki yaşında meme karsinomu, 38 yaşında beyinde glioblastoma multiforme ve ardından 1 ay sonra retroperitoneal malign fibröz histiyositom gelişen bir hastanın anemnezinde annesinin aynı yaşlarda çok sayıda maligniteler nedeni ile öldüğü biliniyor. Bu hastada ön planda düşünülmesi gereken genetik kanser tablosu hangisidir?
A) Von Hippel Lindau Sendromu
B) Li-Fraumeni Sendromu *****
C) Wolfram Hastalığı
D) Multiple Endokrin Neoplazi
E) Retinoblastom gen hasarı
Yanıt – B
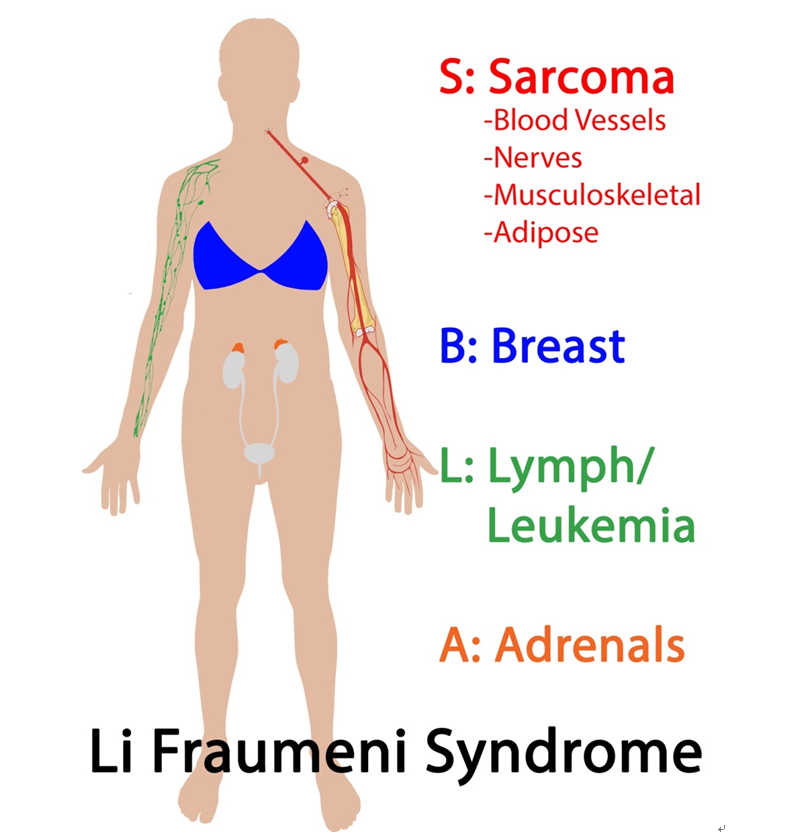
Li-Fraumeni sendromu: 50 yaşın altında oldukça farklı multipl tümör gelişimini (sıklıkla 3 farklı tümör) tanımlar. En sık çıkan tümörler yumuşak doku sarkomları, kolon karsinomu, osteosarkom, meme kanseri, lösemi, beyin tümörleri, adrenal korteks kanseridir. Tanı için, hastada 3 farklı primer kanser olmalı ve en az biri 45 yaş altında saptanmalıdır ve hastanın birinci derece akrabalarından birinde 45 yaş altında benzer karakterde tümör gelişimi bulunmalıdır.
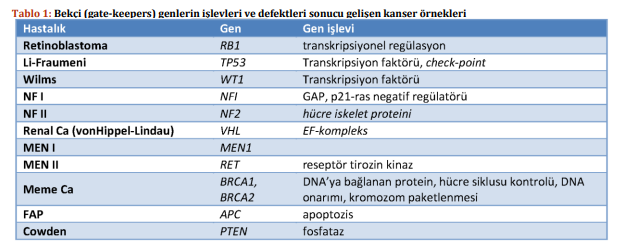
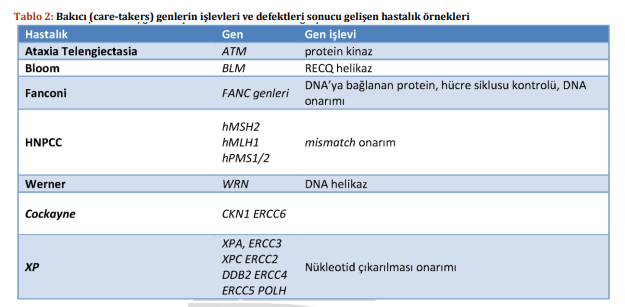
p53 gen (önemli bir tümör süpresör molekül olup genomun gardiyanı olarak da adlandırılır) ürünü olan protein, DNA hasarı ya da hipoksi durumlarında aktifleşir ve DNA’ya bağlanır (bu hasarlı hücrede apopitozun başlangıcıdır). Bu hücrede p53’ün aktivasyonu ile p21 (CDK -sikline bağlı kinaz- inhibitörü) üretilir ve bu hücreyi G1’de durdurur. Mitozu durdurulan hücrede üretilen bcl-2 ile uyarılan GADD45 (DNA onarımından sorumlu) DNA’yı onarmaya çalışır. Eğer onarım sağlanamaz ise (DNA onarılır ise hücrede p53 yıkılır ve hücre normal yaşamına geri döner) bax (apopitozda hücreyi ölüme götüren gen) aktif hale geçer ve hücre apopitoz ile yok olur. DNA hasarı gerçekleşen hücrede p53 geni mutasyon nedeni ile nonaktive ise hücre siklüsü durmayacak ve DNA onarımı yapılamayacaktır ve bu mutant hücre bozuk DNA’sı ile mitoza girdiğinde DNA’da ek mutasyonlarında eklenmesi ile malignité yönünde değişim oluşma olasılığı çok artacaktır.
Li-Fraumeni sendromunda hasta kişide doğumda tüm vücut hücrelerinde 17. kromozomunda bulunan p53 geni hasarlıdır. Sonuçta hastada erken yaşta multipl maligniteler gelişir.
Sporadik olarak gelişen meme ve kolon karsinomu gibi malignitelerde ise malign hücrelerin kontrolsüz bölünmeleri esnasında, malign tümör oluşumu sonrasında sadece malign hücrelerde p53 geni yıkılır; bu o tümör için agresif gidişe neden olabilecek bir değişikliktir. P53 mutasyonu malignitenin agresifliğini arttırır ve sık rastlanan bir özelliktir (meme ve kolon karsinomlarında %80 p53 bozulur). Bütün insan maligniteleri söz konusu olduğunda en sık saptanan genetik bozukluk p53 genin- dedir.
Diğer şıklardaki tablolardan hiç birinde Li-Fraumeni sendromundaki gibi yaygın ve çok sayıda malign tümörün birlikteliği yoktur.
Li-fraumeni sendromu
17. kromozomun kısa koluna yerleşen tümör süpressör genlerden biri olan p53teki bozukluk sonucu görülür. Kolon,meme,akciğer kanserleri ile lösemi,lenforma,osteosarcoma,beyin tümörü gelişir.
Tümör supresör p53 genindeki mutasyona bağlı olarak,bazı kanser türlerine karşı yatkınlık artışı ile karakterize ailesel genetik bir hastalıktır.
————————————-
1-LFS1: TP53’teki Mutasyonlar
Normal koşullar:TP53, normal olarak normal hücre döngüsü üzerindeki etki yoluyla hücre bölünmesinin ve büyümesinin kontrolüne yardımcı olan, kromozom 17 üzerindeki bir tümör baskılayıcı gendir . TP53 tipik olarak DNA hasarı gibi hücresel stresörler nedeniyle eksprese edilir ve onarılabilir DNA hasarının onarımına yardımcı olmak için hücre döngüsünü durdurabilir veya onarılamaz hasara sahip bir hücrenin apoptozunu indükleyebilir. “Kötü” DNA’nın onarımı veya bir hücrenin apoptozu, hasarlı hücrelerin çoğalmasını önler.
Mutant koşullar: TP53’ün mutasyonları , normal işlevini engelleyebilir ve hasarlı DNA’ya sahip hücrelerin bölünmeye devam etmesine izin verebilir . Bu DNA mutasyonları kontrol edilmezse, bazı hücreler kontrolsüz bir şekilde bölünerek tümörler (kanserler) oluşturabilir. DNA’daki diğer mutasyonlar, vücudun farklı bölgelerine seyahat edebilen ve kanser geliştirebilen habis hücrelere yol açabilir. Li-Fraumeni sendromlu birçok kişinin bir TP53 mutasyonu için heterozigot olduğu gösterilmiştir . Son araştırmalar, klasik LFS ailelerinin %60 ila %80’inin , çoğunluğu DNA bağlama alanındaki yanlış anlamlı mutasyonlar olan saptanabilir germ-line TP53 mutasyonlarını barındırdığını göstermiştir . Bu yanlış anlamlı mutasyonlar, p53’ün DNA’ya bağlanma yeteneğinde bir azalmaya neden olarak normal TP53 mekanizmasını inhibe eder.

Eşsiz Brezilya mutasyonu: Li–Fraumeni sendromuna yol açan diğer mutasyonlar DNA bağlama alanı dışında bulunmuş olsa da, TP53’ün tetramerizasyon alanının kodon 337’sindeki bir mutasyon özellikle yüksek bir frekans göstermiştir. Tetramerizasyon alanı, bir tetramer olarak bulunan p53 proteininin oligomerizasyonunda önemli bir rol oynar . Bu mutasyon sadece Brezilyalı ailelerde bulunmuştur ve TP53 geninin 10. eksonunda yer almaktadır. Mutasyon, kodon 337’de arginin’den histidine bir amino asit değişimine neden olur. Düşük ila normal fizyolojik aralıkta (7.5’e kadar) pH ile, mutant protein normal oligomerler oluşturur ve baskılayıcı işlevini korur. Bununla birlikte, yüksek bir fizyolojik pH’ta, p53, bir tetramer halinde birleşemez. Bu benzersiz özellik, bu özel mutasyona sahip ailelerin neden genellikle eksik penetrasyon gösterdiğine katkıda bulunabilir.

Dominant negatif mutasyonlar: Li–Fraumeni sendromlu çoğu birey, bir mutant TP53 geni için heterozigottur ve bazı p53 mutantları, vahşi tip p53’ün işlevini baskın bir negatif şekilde inhibe edebilir. Mutasyona uğramış p53 proteinleri tipik olarak vahşi tipten daha kararlıdır ve vahşi tip proteinin hücre proliferasyonunu baskılama ve hücre döngüsü durmasını tetikleme aktivitesini inhibe edebilir. Mutant p53’ün bazı vahşi tip p53’ü inhibe edebilmesi nedeniyle, hasarlı hücreler çoğalmaya ve dönüşmeye daha da yatkındır ve bu da kansere neden olur.
2-LFS2: CHEK2’deki mutasyonlar
Li-Fraumeni’nin biraz tartışmalı olan başka bir varyantı, CHEK2 (veya CHK2 ) geninin bir mutasyonudur . CHK2 ayrıca bir tümör baskılayıcı gendir ; p53’ün eylemini düzenler ve DNA hasarını tespit eden ATM tarafından etkinleştirilir ve bu şekilde, DNA hasarı bilgisi, DNA onarımının gerçekleşebilmesi için o noktada hücre döngüsünü dolaylı olarak durdurmak üzere p53’e iletilebilir. apoptoza (programlanmış hücre ölümü) neden olur.
3-LFS-L :
Klasik Li – Fraumeni sendromunun kriterlerine uymayan aileler “LFS benzeri” olarak adlandırılmıştır LFS benzeri bireylerde genellikle saptanabilir herhangi bir p53 mutasyonu yoktur ve Birch veya Eeles kriterlerine göre teşhis konulabilir.
Üçüncü bir lokus, 1. kromozomun uzun koluna (1q23) eşlendi , ancak henüz bir gen tanımlanmadı.
Bu sendromla ilişkilendirilen başka bir lokus, CDKN2A – CDKN2B’dir .

Li-Fraumeni sendromu (LFS), çok çeşitli çocukluk ve yetişkinlik başlangıçlı maligniteler için yüksek risklerle ilişkili bir kansere yatkınlık sendromudur. LFS’li bireylerde yaşam boyu kanser riski erkekler için ≥% 70 ve kadınlar için ≥% 90’dır. Beş kanser türü, LFS tümörlerinin çoğunu oluşturur: adrenokortikal karsinomlar, meme kanseri, merkezi sinir sistemi tümörleri, osteosarkomlar ve yumuşak doku sarkomları.
LFS, lösemi, lenfoma, gastrointestinal kanserler, baş ve boyun kanserleri, böbrek, gırtlak, akciğer, deri (örn. Melanom), yumurtalık, pankreas, prostat, testis ve tiroid dahil olmak üzere birçok ek kanser riskinde artış ile ilişkilidir. LFS’li bireyler, çocuklukta ve genç yetişkinlikte kanser için yüksek risk altındadır; Hayatta kalanlar, birden fazla birincil kanser için yüksek risk altındadır. Li-Fraumeni sendromlu kişilerde diğer bazı kanser türleri de daha sık görülür .
Li-Fraumeni sendromunun dünya çapında 5.000 ila 20.000 kişide 1’e ortaya çıktığı düşünülmektedir.
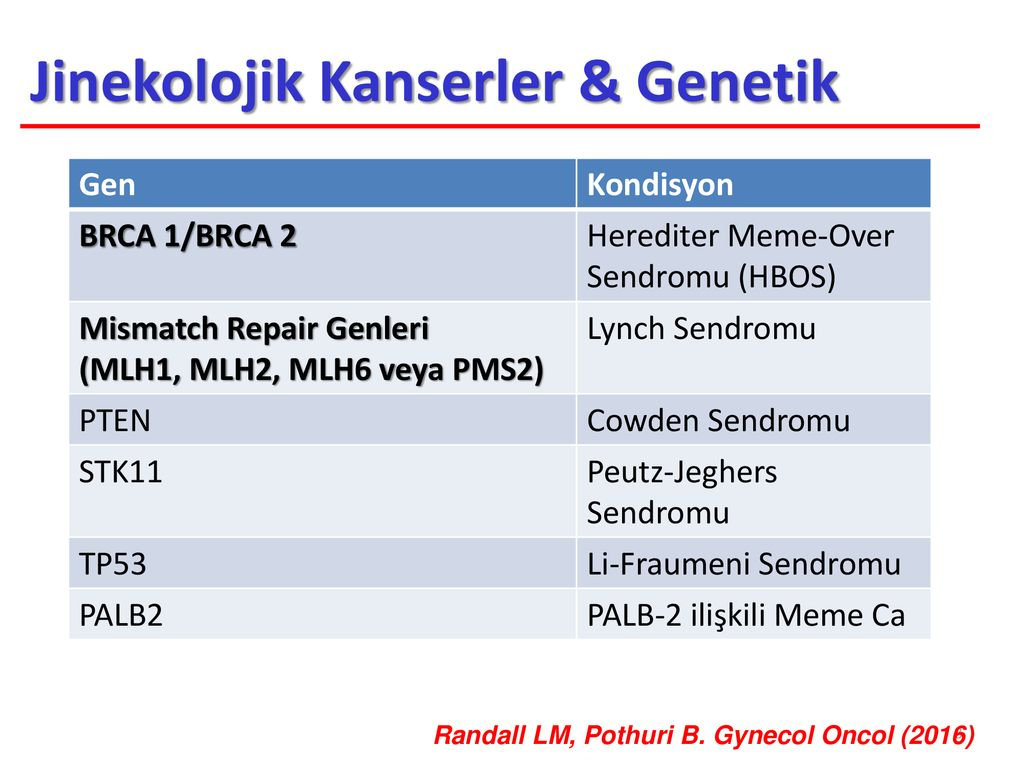
Li-Fraumeni sendromu , TP53 genindeki mutasyonlarla ilişkilidir . Li-Fraumeni sendromlu ailelerin yaklaşık dörtte üçünde TP53 geninde germ hattı mutasyonları vardır.

Germline mutasyonları tipik olarak kalıtsaldır ve vücuttaki her hücrede TP53 genin bir kopyasında mutasyon mevcuttur.
TP53 tümör baskılayıcı bir gendir, yani normalde hücrelerin büyümesini ve bölünmesini kontrol etmeye yardımcı olur.
Bu gendeki mutasyonlar, hücrelerin kontrolsüz bir şekilde bölünmesine ve tümör oluşturmasına izin verebilir. TP53 mutasyonları olan kişilerde diğer genetik ve çevresel faktörlerin de kanser riskini etkilemesi muhtemel gözükmektedir.
Li-Fraumeni sendromuna ve Li-Fraumeni benzeri sendroma özgü kansere sahip birkaç ailede TP53 mutasyonları yoktur. Bu vakalarda yer alan genetik faktörler belirsizdir.
Li-Fraumeni sendromu , otozomal dominant bir modelde kalıtılır; bu, her hücrede değiştirilmiş genin bir kopyasının kanser geliştirme riskini artırmak için yeterli olduğu anlamına gelir.
Li-Fraumeni sendromlu çoğu insan, etkilenen bir ebeveynden genin değiştirilmiş bir kopyasını miras alır.
Vakaların 7 ila 20 oranında ise, değişmiş gen bir sonucudur yeni (de novo) mutasyonu geni üreme hücrelerinin (yumurta veya sperm) oluşumu sırasında veya gelişimin çok erken döneminde meydana gelir.
Li-Fraumeni sendromunda bir kanserin gelişmesi için, bir kişinin yaşamı boyunca vücut hücrelerinde TP53 geninin diğer kopyasını içeren bir mutasyon meydana gelmelidir. Bu genin iki değiştirilmiş kopyasına sahip hücreler, tümörlerin gelişmesine izin veren fonksiyonel TP53 proteini yapmaz.
Bir TP53 gen mutasyonunu miras alan hemen hemen herkes, sonunda bazı hücrelerde genin ikinci kopyasında bir mutasyon kazanacaktır (çift vuruş hipotezi). İkinci mutasyon genellikle meme, kemik veya kas dokusundaki hücrelerde meydana gelir ve tipik olarak Li-Fraumeni sendromunda yaygın olan tümörlere yol açar.
TP53 geni (kromozom 17 p13.1), tümör proteini, p53 (veya p53) olarak adlandırılan bir proteinin yapmak için talimatlar içerir. Bu protein, bir tümör baskılayıcı görevi görür; bu hücrelerin çok hızlı veya kontrolsüz bir şekilde büyümesini ve bölünmesini (çoğalmasını) önleyerek hücre bölünmesini düzenlediği anlamına gelir. P53 proteini, DNA onarımını ve hücre bölünmesini düzenlemek için gerekli olduğundan, ” TP53 geni genomun koruyucusu” yani genomun gardiyanı gibi işlev görür.
P53 proteini, doğrudan DNA’ya bağlandığı (bağlandığı) vücuttaki hücrelerin çekirdeğinde bulunur. Bir hücredeki DNA, güneş ışığından kaynaklanan toksik kimyasallar, radyasyon veya ultraviyole (UV) ışınları gibi maddeler tarafından hasar gördüğünde, bu protein, DNA’nın onarılıp onarılmayacağını veya hasarlı hücrenin kendi kendini yok edip etmeyeceğini belirlemede kritik bir rol oynar apoptoz). DNA onarılabilirse, p53 hasarı düzeltmek için diğer genleri etkinleştirir. DNA onarılamazsa, bu protein hücrenin bölünmesini önler ve apoptoza girmesi için sinyal verir. p53 mutasyona uğramış veya hasar görmüş DNA’ya sahip hücrelerin bölünmesini durdurarak tümör gelişimini önlemeye yardımcı olur.
LFS, otozomal dominant bir şekilde miras alınır. LFS teşhisi konan çoğu birey, bir ebeveynden TP53 patojenik bir varyantı miras almıştır. De novo germline TP53 patojenik varyantına sahip bireylerin oranının % 7 ile % 20 arasında olduğu tahmin edilmektedir. LFS tanısı alan bir bireyin (yani, klasik LFS kriterlerini karşılayan ve / veya heterozigot germ hattı TP53 patojenik varyantına sahip bir birey).
LFS’ye neden olan patojenik bir varyantı kalıtım yoluyla alma ve ilişkili kanser risklerine sahip olma riski% 50’dir. LFS bakımından risk altındaki aile üyeleri için öngörücü testler , doğum öncesi testler ve ailede bir TP53 germ hattı patojenik varyantı tanımlanmışsa implantasyon öncesi genetik test mümkündür .
Li-Fraumeni benzeri sendromda ise, 22q12 kromozomundaki CHEK2(checkpoınt kinase 2) genindeki heterozigot mutasyona mutlaka bakılmasında fayda vardır.
Aşağıdaki maddelerden birisinin varlığı TP53 testi için endikasyon oluşturur
1) Ailede TP53 mutasyonu olan birey varlığı.
2) Li-Fraumeni kriteri olan kombinasyonun varlığı
a) 45 yaş altında sarkom tanısı almış olmak
b) En az bir birinci derece yakın akrabanın 45 yaş altında kanser tanısı alması
c) Buna ek olarak ailenin aynı tarafından bir 1. ya da 2. derece yakın akrabanın herhangi bir yaşta sarkom tanısı alması ya da 45 yaş altında kanser tanısı alması.
3) Li-Fraumeni benzeri sendrom kriteri olan kombinasyonun varlığı
a) 45 yaş altında çocukluk çağı sarkomları, beyin tümörü, adrenokortikal karsinom tanısı almış olmak
b) En az bir birinci ya da ikinci derece yakın akrabanın herhangi bir yaşta tipik Li-Fraumeni tümörü tanısı alması
c) Buna ek olarak ailenin aynı tarafından bir 1. ya da 2. derece yakın akrabanın 60 yaş altında kanser tanısı alması
4) 30 yaş altında meme kanseri tanısı aldığı halde BRCA1/BRCA2 mutasyonu negatif çıkan ve özellikle aile öyküsünde sarkom, beyin tümörü veya adrenokortikal karsinom olan hastalar.