
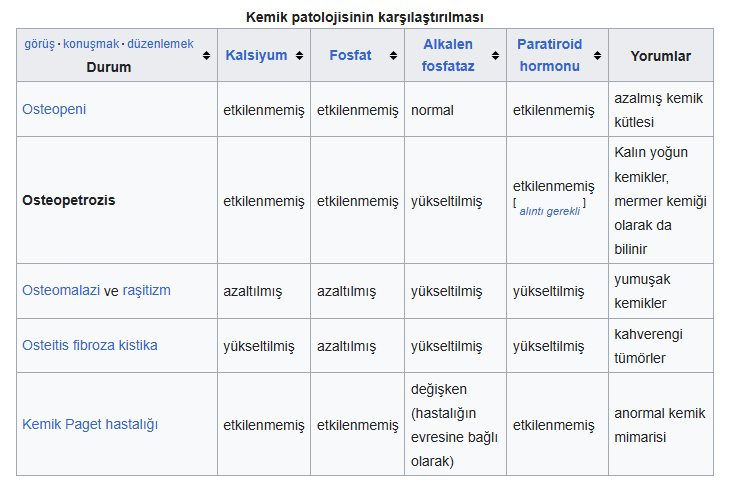
Kemik içinde kemik görünümü, yaygın dansite artışı, foramenlerde daralma, kemik medullasının kaybolması
gibi özellikler hangi hastalığın özellikleridir?
A) Piknodisostozis
B) Osteopetrozis
C) Osteogenezis imperfekta
D) Vitamin D intoksikasyonu
E) Hipoparatiroidizm
CEVAP: B
Osteopetrosis gelişme geriliği, burun tıkanıklığı, merkezi sinir sistemi bulguları, hepatosplenomegaliyle seyreden artmış kemik dansitesiyle karakterize bir hastalıktır.
Osteoklastların kemik rezorbsiyonundaki yetersizliği sonucu olduğu düşünülmektedir.
Otozomal dominant forma ve otozomal resesif forma sahiptir. Hematolojik olarak kemik iliği aşırı kemik gelişimi nedeni ile progresif olarak daralır.
Progresif pansitopeni görülür. Kemik iliği hipoplazisi oluşur. Kemik iliği aspirasyonunun zoluğu anlamlı olsada tanı için şart değildir.
Kompansatuar ekstrameduller hematopoez görülür.
Radyolojik bulgular önemlidir. Artmış kemik dansitesi en önemli bulgudur. Metafiz bölgesindeki kemikleşme bozukluğu kemik içinde kemik görüntüsü verir. Kraniyografide orbita çevresinde gözlük şeklinde dansite artışı vardır. Özelikle uzun kemiklerde fraktür görülebilir.
Tedavide steroid, kalsitriol, vit D3, eritropoetin, gama interferon kullanılmakla birlikte kesin tedavisi kemik iliği transplantasyonudur.
Hastalığın prognozu tedavisiz olgularda kötüdür.
Anemi, kanama ve enfeksiyona sekonder olarak ölüm sıktır.
Büyüme gelişme geriliği morbiditeyi arttırır.
2 tipi vardır
a-Malign infantil osteopetrozis
Tipik olarak bebeklik döneminde ortaya çıkan ve yaygın hiperostozisin (aşırı kemik büyümesi) benzersiz radyografik görünümü ile karakterize, nadir görülen bir osteosklerozan iskelet displazisi türüdür .
Kemik yoğunluğundaki genel artış, kortekslerin nispeten korunduğu medüller kısmı içermeye yönelik özel bir yatkınlığa sahiptir. Kemik iliği boşluklarının obliterasyonu ve buna bağlı hücresel fonksiyonun baskılanması ciddi hematolojik komplikasyonlara yol açabilir. Kemik genişlemesine bağlı optik atrofi ve kranial sinir hasarı belirgin morbiditeye yol açabilir. Tedavi edilmeyen vakalarda prognoz son derece kötüdür. Düz radyografi tanıya yönelik temel bilgileri sağlar. Klinik ve radyolojik korelasyonlar da tanı süreci için temeldir ve ek gen testleri doğrulayıcıdır.
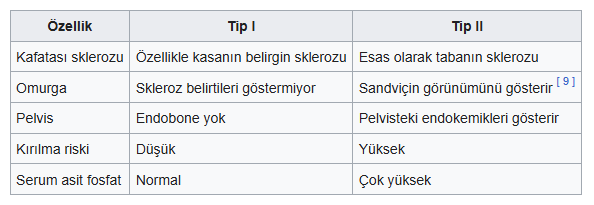
b-Erişkin osteopetrozisi
Otozomal dominant osteopetroz (ADO), Albers-Schönberg hastalığı olarak da bilinir. Çoğu kişi bu bozukluğa sahip olduğunu bilmez çünkü çoğu birey herhangi bir semptom göstermez. Ancak, semptom gösterenlerde genellikle omurga eğriliği ( skolyoz ) ve çoklu kemik kırıkları görülür. Radyografik, biyokimyasal ve klinik özelliklere göre iki tür yetişkin osteopetrozu vardır.
ADO’nun Tip I ve Tip II şeklinde 2 subtipi vardır.
Normal kemik büyümesi, osteoblastlar tarafından kemik oluşumu ve osteoklastlar tarafından kemik rezorpsiyonu (kemik matrisinin parçalanması) arasındaki denge ile sağlanır. Osteopetrozda, osteoklast sayısı azalabilir, normal olabilir veya artabilir. En önemlisi, osteoklast disfonksiyonu bu hastalığın patogenezine aracılık eder.
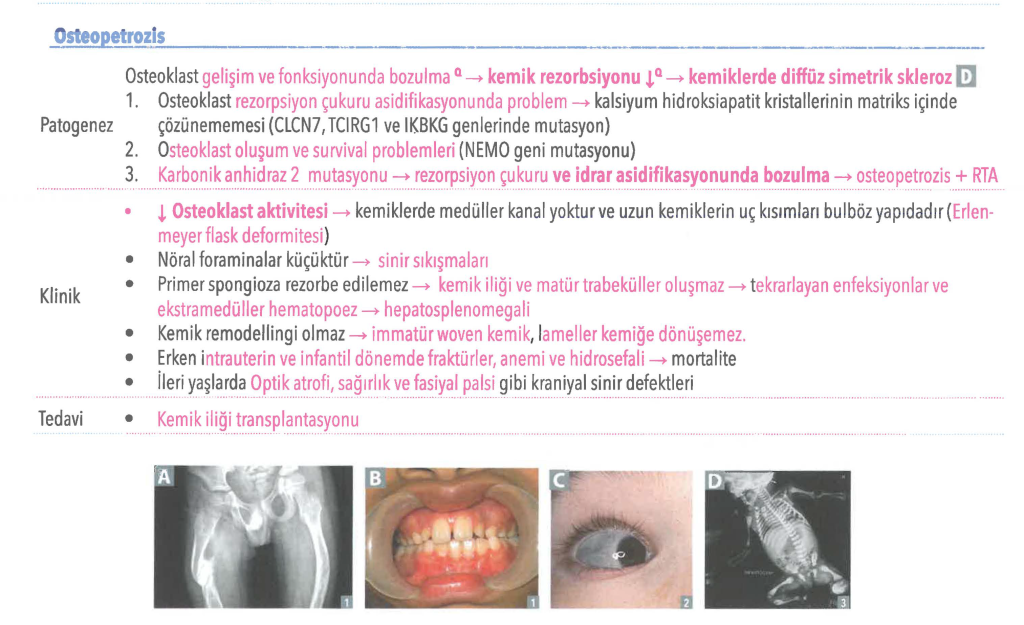
Osteopetroz, CA2 geni tarafından kodlanan karbonik anhidraz enziminin eksikliği gibi osteoklast rezorpsiyon çukurunun asitlenmesine müdahale eden altta yatan mutasyonlar nedeniyle oluşabilir. Karbonik anhidraz, osteoklastlar tarafından proton üretimi için gereklidir. Bu enzim olmadan hidrojen iyon pompalaması engellenir ve osteoklastlar tarafından kemik rezorpsiyonu kusurludur, çünkü kalsiyum hidroksiapatiti kemik matrisinden ayırmak için asidik bir ortama ihtiyaç vardır. Kemik oluşumu devam ederken kemik rezorpsiyonu başarısız olduğunda, aşırı kemik oluşur.
Osteopetrozisle ilişkili genlerden herhangi birindeki mutasyonlar anormal veya eksik osteoklastlara yol açar. İşlevsel osteoklastlar olmadan, yeni kemik oluşurken eski kemik parçalanmaz. Sonuç olarak, iskelet boyunca kemikler alışılmadık şekilde yoğunlaşır. Kemikler ayrıca yapısal olarak anormaldir ve bu da onları kırılmaya yatkın hale getirir. Kemik yeniden şekillenmesindeki bu sorunlar osteopetrozisin tüm önemli özelliklerinin altında yatar.
İnterferon gama-1b, şiddetli, kötü huylu osteopetrozlu hastalarda hastalığın ilerleme süresini geciktirmek için FDA tarafından onaylanmışt
———————-
Aşağıdakilerden hangisi Osteopetrozis patogenezinde rol alan genetik mutasyonlardan değildir?
a-CLCN/
b-TCIRG1
c-IKBKG
d-FGF23 ***
e-NEMO
————————
Osteopetroz (Mermer Kemik Hastalığı / Albers-Schönberg Hastalığı), nadir görülen, genetik bir kemik hastalığıdır. Bu hastalıkta, kemiklerin normalden daha yoğun ve kırılgan olmasına neden olan osteoklast disfonksiyonu (kemik yıkımından sorumlu hücrelerin işlev bozukluğu) temel patolojiyi oluşturur.
Normal Kemik Remodelling (Yeniden Şekillenme): Sağlıklı bir kemik dokusunda, iki ana hücre türü denge içinde çalışır:
- Osteoblastlar: Yeni kemik dokusu oluşturur.
- Osteoklastlar: Eski ve hasarlı kemik dokusunu yıkar (rezorpsiyon).
Osteopetrozda, osteoklastlar ya yeterli sayıda değildir ya da işlevlerini düzgün bir şekilde yerine getiremezler. Bu durum, kemik yıkımının (rezorpsiyon) yetersiz kalmasına ve kemik oluşumunun (osteoblast aktivitesi) baskın gelmesine yol açar. Sonuç, aşırı yoğun, sklerotik (sertleşmiş) ancak paradoksal olarak kırılgan kemiklerdir.
Etiyoloji (Nedenleri):
Osteopetroz, genetik heterojenite gösterir; yani birçok farklı genin mutasyonu bu hastalığa neden olabilir. En yaygın mutasyonlar şunlardır:
- CLCN7 geni: En sık mutasyonlardan biridir ve klorid kanalı işlevini etkiler.
- TCIRG1 geni: Osteoklastların asit ortamı oluşturma yeteneğini bozan bir proton pompasını kodlar.
- CA2 geni: Karbonik anhidraz II enzimini kodlar ve kemik rezorpsiyonunda rol oynar.
- Kalıtım şekli, mutasyonun olduğu gene ve şiddetine bağlı olarak otozomal resesif (daha şiddetli formlar) veya otozomal dominant (daha hafif formlar) olabilir.
Sınıflandırma ve Klinik Belirtiler:
Osteopetroz, şiddetine ve genetik geçişine göre farklı tiplere ayrılır:
- Malign İnfantil Osteopetroz (Otozomal Resesif Osteopetroz – ARO):
- En şiddetli formdur ve genellikle bebeklik veya erken çocukluk döneminde ortaya çıkar.
- Belirtiler:
- Kemik İliği Yetmezliği: Kemiklerin aşırı yoğunlaşması nedeniyle kemik iliği boşluğu daralır. Bu durum, anemi (kansızlık), trombositopeni (düşük trombosit sayısı) ve lökopeni (düşük beyaz kan hücresi sayısı) gibi hematolojik sorunlara yol açar. Enfeksiyonlara ve kanamalara yatkınlık artar.
- Sinir Sıkışmaları: Kafatası kemikleri kalınlaştığı için kraniyal sinirler (optik sinir, işitsel sinir vb.) sıkışabilir. Bu durum, körlük, sağırlık, yüz felci gibi nörolojik bozukluklara neden olabilir.
- Kemik Kırıkları: Kemikler yoğun olmasına rağmen, esnekliğini kaybettiği için paradoksal olarak kolayca kırılır (kemik kırılganlığı).
- Büyüme Geriliği: Genel gelişimsel gerilik.
- Diş Anomalileri: Dişlerde gelişimsel bozukluklar.
- Hidrosefali: Beyinde omurilik sıvısı birikimi.
- Prognoz: Tedavi edilmezse, yaşamın ilk on yılında, genellikle enfeksiyonlar veya kanama komplikasyonları nedeniyle ölümle sonuçlanabilir.
- İntermediate Osteopetroz (Ara Form):
- Hem otozomal resesif hem de otozomal dominant geçiş gösterebilir.
- Belirtiler infantildeki kadar şiddetli değildir, ancak yine de anemi, sinir sıkışmaları ve kırıklar görülebilir.
- Tanı genellikle çocukluk veya ergenlik döneminde konur.
- Benign Otozomal Dominant Osteopetroz (ADO) / Albers-Schönberg Hastalığı:
- En hafif formdur ve genellikle asemptomatiktir veya hafif semptomlarla seyreder.
- Genellikle tesadüfen, başka bir nedenle çekilen röntgenlerde veya patolojik kırıklar sonrasında fark edilir.
- Belirtiler: En sık patolojik kırıklar ve bazen hafif anemi veya sinir sıkışmaları görülür.
- Prognoz: Genellikle normal yaşam süresine sahiptirler.
Tanı:
- Radyografi (Röntgen): Kemiklerin genel yoğunluğundaki artış, özellikle uzun kemiklerdeki “kemik içinde kemik” görünümü (endobon içinde endobon) veya omurlardaki “sandviç” görünümü karakteristiktir.
- Kan Testleri: Anemi, lökopeni, trombositopeni gibi hematolojik anormallikler görülebilir. Kalsiyum, fosfat ve paratiroid hormon seviyeleri genellikle normaldir, bu da diğer kemik hastalıklarından ayırımda önemlidir.
- Genetik Testler: Kesin tanı için mutasyon analizi yapılır.
- Kemik Biyopsisi: Nadiren gerekse de, histolojik olarak osteoklast fonksiyon bozukluğu gösterilebilir.
Tedavi:
Tedavi, hastalığın tipine ve şiddetine bağlıdır.
- Malign İnfantil Osteopetroz (ARO) için:
- Hematopoetik Kök Hücre Nakli (HSCT): Tek küratif (iyileştirici) tedavi seçeneğidir. Başarılı nakil, işlevsel osteoklastların oluşmasını sağlar ve kemik iliği yetmezliği ile sinir sıkışmalarını iyileştirebilir.
- İnterferon gama-1b: Bazı durumlarda kemik rezorpsiyonunu artırmaya yardımcı olabilir.
- Kortikosteroidler: Anemiyi geçici olarak düzeltmek için kullanılabilir.
- Diyet: Düşük kalsiyum, yüksek fosfatlı diyetler denenebilir.
- Daha Hafif Formlar (ADO):
- Genellikle semptomatik tedavi ve komplikasyonların yönetimi (kırıkların ortopedik tedavisi, sinir sıkışmaları için dekompresyon cerrahisi).
- Bazı durumlarda D vitamini ve kalsiyum takviyeleri kullanılabilir.
Osteopetroz, nadir bir hastalık olmasına rağmen, erken tanı ve özellikle şiddetli formlarda erken kök hücre nakli, hastaların prognozunu önemli ölçüde iyileştirebilir.
Osteopetroz, temelinde osteoklastların kemik rezorpsiyonu (yıkımı) işlevindeki genetik bir bozukluğun yattığı heterojen bir hastalıktır. Bu bozukluk, kemiklerin aşırı yoğunlaşmasına ve bir dizi klinik soruna yol açar. Hastalığın genetik yapısı oldukça karmaşıktır, çünkü birçok farklı genin mutasyonu osteopetroza neden olabilir ve kalıtım modeli mutasyona uğrayan gene göre değişebilir.
İşte osteopetroz ile ilişkili başlıca genetik mutasyonlar ve bunların yol açtığı fenotipler:
Ana Patofizyoloji: Tüm osteopetroz tiplerinde ortak olan patoloji, osteoklastların asit ortam oluşturma ve/veya kemiği sindirme yeteneğindeki bozukluktur. Osteoklastlar, kemik yüzeyine tutunarak bir sızdırmazlık bölgesi oluşturur ve bu bölgeye proton (H+) ve klor (Cl-) iyonları salgılayarak kemiği çözen asidik bir mikroçevre oluşturur. Daha sonra enzimleri (katepsin K gibi) salgılayarak organik matriksi sindirirler. Osteopetroza neden olan gen mutasyonları genellikle bu süreçlerden birini veya birkaçını aksatır.
Başlıca Genler ve Kalıtım Modelleri:
- TCIRG1 Geni (T-cell, immune, regulatory, 1, H+ transporting V1 subunit A3):
- Kalıtım: Genellikle otozomal resesif geçişli (malign infantil osteopetrozun en yaygın nedeni).
- İşlev: Bu gen, osteoklastlarda bulunan ve kemik matriksini asitlendirmek için protonları (H+) dışarı pompalayan v-ATPaz adı verilen bir proton pompasının alt birimini kodlar.
- Mutasyon Etkisi: TCIRG1 mutasyonları, işlevsel bir proton pompasının oluşmasını engelleyerek osteoklastların kemiği düzgün bir şekilde asitlendirememesine ve dolayısıyla rezorbe edememesine neden olur. Bu, tipik olarak şiddetli, erken başlangıçlı (malign infantil) osteopetroza yol açar.
- CLCN7 Geni (Chloride Channel 7):
- Kalıtım: Hem otozomal resesif (şiddetli form) hem de otozomal dominant (daha hafif form, Albers-Schönberg hastalığı) geçişli mutasyonlar görülebilir.
- İşlev: Bu gen, osteoklastların işlevi için kritik olan bir klorid kanalını kodlar. Klorid kanalı, proton pompası ile birlikte çalışarak hücre dışına proton salgılanması sırasında elektriksel dengeyi sağlar.
- Mutasyon Etkisi: CLCN7 mutasyonları, klorid kanalının işlevini bozarak osteoklastların yeterince asit salgılayamamasına neden olur. Otozomal resesif mutasyonlar daha şiddetli bir fenotip gösterirken, otozomal dominant mutasyonlar genellikle daha hafif bir klinik tabloya (ADO) yol açar. Bu, ADO’nun en yaygın genetik nedenidir.
- CA2 Geni (Carbonic Anhydrase 2):
- Kalıtım: Otozomal resesif geçişli.
- İşlev: Bu gen, hücre içinde proton (H+) ve bikarbonat (HCO3-) üreten karbonik anhidraz II enzimini kodlar. Osteoklastlar, kemiği çözmek için gereken protonları bu enzim aracılığıyla üretir.
- Mutasyon Etkisi: CA2 mutasyonları, bu enzimin eksikliğine veya işlevsizliğine yol açar. Bu durum, osteoklastların yeterli proton üretememesine ve dolayısıyla kemiği rezorbe edememesine neden olur. Bu tip osteopetroz, renal tübüler asidoz ve serebral kalsifikasyonlar gibi ek bulgularla karakterize olabilir.
- OSTM1 Geni (Osteopetrosis Associated Transmembrane Protein 1):
- Kalıtım: Otozomal resesif geçişli.
- İşlev: Bu genin tam işlevi henüz tam olarak anlaşılamamıştır, ancak osteoklast fonksiyonu için önemli olduğu düşünülmektedir.
- Mutasyon Etkisi: Mutasyonları, şiddetli infantilde görülebilir ve sıklıkla beyin gelişimi anomalileri ve bağışıklık sistemi sorunları (bazen immun yetmezlik) ile ilişkilidir.
- SNX10 Geni (Sorting Nexin 10):
- Kalıtım: Otozomal resesif geçişli.
- İşlev: Endozomal trafik ve vezikül oluşumunda rol oynadığı düşünülmektedir, bu da osteoklast fonksiyonu için önemlidir.
- Mutasyon Etkisi: Mutasyonları, şiddetli osteopetroza neden olur.
- PLEKHM1 Geni (Pleckstrin Homology Domain Containing A1):
- Kalıtım: Otozomal resesif geçişli.
- İşlev: Osteoklastların lizozomal fonksiyonları ve kemik rezorpsiyonu için kritik olan bir protein kodlar.
- RANKL/RANK/OPG Yolu İlişkili Genler (Daha Nadir):
- Kemik remodellingini düzenleyen bu yolaktaki genlerdeki (örn. TNFRSF11A [RANK] mutasyonları) nadir mutasyonlar da bazı osteopetroz formlarına neden olabilir.
Genetik Tanı ve Klinik Korelasyon:
- Osteopetroz tanısı alan bir hastada, genetik testler, spesifik mutasyonu tanımlamak ve hastalığın kalıtım modelini belirlemek için hayati öneme sahiptir.
- Genetik tanı, hastalığın şiddetini (fenotipini) tahmin etmede ve tedavi stratejilerini (özellikle kök hücre nakli gerekliliği) belirlemede yardımcı olur.
- Örneğin, TCIRG1 mutasyonları genellikle acil kök hücre naklini gerektiren en şiddetli formu işaret ederken, CLCN7 mutasyonları (ADO) genellikle daha hafif bir seyir gösterir.
Osteopetrozun genetik heterojenitesi, hastalığın patofizyolojisinin karmaşıklığını ve bireyselleştirilmiş tanı ve tedavi yaklaşımlarının önemini vurgular.
Aşağıdaki kemik patolojilerinden hangisi enzim eksikliğine bağlı gelişir?
A) Osteopetrozis
B) Paget hastalığı
C) Brown tümör
D) Akondroplazi
E) Fibröz displazi
CEVAP:A
Osteopetrozis hastalığında Osteoklast aktivitesinde defekt vardır. Karbonik anhidraz II defektine bağlıdır (asidifikasyon bozuk). Kemikle diffüz skleroz nedeniyle mermer gibi sert ancak tebeşir gibi kolay kırılır. Grafide çok radyodens görüntü izlenir.Daralan deliklerde sinir sıkışmasına bağlı kranial sinir felçleri görülür. Pansitopeni görülür. Bu nedenle ekstramedüller hematopoez gelişir (karaciğer ve dalak). Anemi, Tekrarlayan enfeksiyonlar ve Hepatosplenomegali görülür.