
Nöromiyelitis optika ( NMO ) dahil olmak üzere nöromiyelitis optika spektrum bozuklukları ( NMOSD), optik sinirin ( optik nörit , ON) ve omuriliğin ( miyelit ) akut inflamasyonu ile karakterize edilen otoimmün hastalıklardır.
Devic hastalığı multipl skleroza oldukça benzeyen bir otoimmün tabanlı nörolojik hastalıktır, bu hastalıkta, tıpkı ms’te olduğu gibi vücudun bağışıklık sistemi optik sinire ve omuriliğe saldırır.
MS den tek farkı, saldıranın t lenfositleri değil “nöromiyelit optica immunoglobulin g” isimli antibodyleri olmasıdır.
Kesin tanısı 2 alt semptomun bulunmasına bağlıdır: optik nevrit ve akut transvers miyelit, bunun için radyolojik yöntemler de tanı koyulmasında kullanılır.
Hastalık , ms’e benzer şekilde ataklarla ilerler, ataklar ms’te olduğu gibi çoğunlukla relapsing-remitting şeklindedir ve ataklar hastanın görüşünü veya hareketini geri dönüşü olmayacak şekilde engelleyebilir.
Yine tıpkı ms’te olduğu gibi günümüzde atakları ve hastalığın ilerleyişini engelleyici tedavi mümkündür ms’ten farklı olarak interferon yerine bağışıklık sistemini baskılayıcı ilaçlar kullanılmaktadır.

Atakların tedavisinde steroid tedavisinin yanında plazmaferezin de yararlı olduğu bildirilmiştir.
ON ve miyelit atakları eşzamanlı veya ardışık olabilir. Özellikle tedavi edilmeyen hastalarda hastalığın tekrarlayan seyri yaygındır.
Vakaların %80’inden fazlasında NMO’ya, en yaygın olanı olan aquaporin 4’e ( anti -AQP4 ) karşı immünoglobulin G otoantikorları neden olur. Merkezi sinir sistemindeki su kanalı proteini. Anti-AQP4 negatif vakaların bir alt kümesi, miyelin oligodendrosit glikoproteine ( anti-MOG ) karşı antikorlarla ilişkilidir .
Nadiren NMO, diğer otoimmün hastalıklar (örn. bağ dokusu bozuklukları , paraneoplastik sendromlar ) veya bulaşıcı hastalıklar bağlamında ortaya çıkabilir . Bazı durumlarda etiyoloji bilinmemektedir ( idiyopatik NMO).
Multipl skleroz (MS) ve NMO, klinik ve radyolojik görünüm açısından benzer olabilir ve MS, çok nadiren NMO benzeri bir fenotiple ortaya çıkabilir ( örneğin, uzun süreli MS’li hastalarda, uzunlamasına yaygın omurilik lezyonlarını taklit eden birleşik omurilik lezyonlarıyla sonuçlanan hastalarda). tipik olarak NMO’da görülür). Sonuç olarak, NMO geçmişte hatalı bir şekilde MS’in klinik bir varyantı olarak düşünülmüştü. Ancak NMO olguların büyük çoğunluğunda MS ile ilişkili değildir ve patogenez , klinik tablo, manyetik rezonans görüntüleme , beyin omurilik sıvısı bulguları, hastalığın seyri ve prognozu açısından MS’ten önemli ölçüde farklılık göstermektedir.
NMOSD’nin belirti ve semptomları , hastalığın etkilediği nörolojik yapılara ve bir dereceye kadar ilgili antikorlara bağlıdır. Belirti ve semptomlar genellikle tekrarlayan ve düzelen bir seyir izler, ancak bazen ilerleyici (tek fazlı) olabilir. Eksiklikler geçici veya kalıcı olabilir; ikincisi özellikle tedavinin yokluğunda olabilir.
Hastalığın en sık görülen başlangıç belirtisi omurilik iltihabıdır (miyelit). Miyelit, omurilik fonksiyon bozukluğuna neden olur ve bu da kas zayıflığına , uzuvlarda felce, duyu kaybı veya azalmasına, spazmlara, mesane ve bağırsak kontrolünün kaybına veya erektil disfonksiyona neden olabilir. Miyelit enine olabilir , omuriliğin tüm kesitini etkileyebilir ve iki taraflı semptomlar gösterebilir.
Hastalığın ikinci en yaygın başlangıç belirtisi optik sinir ve/veya optik kiazmanın iltihabıdır ( optik nörit , ON). ON, görme keskinliğinde azalma ile değişen derecelerde görme bozukluğuna yol açabilir , ancak görme alanı kusurları veya renkli görme kaybı tek başına veya görme keskinliğinin resmi kaybından önce ortaya çıkabilir. Multipl skleroza (MS) bağlı idiyopatik ON ve ON ile karşılaştırıldığında, NMOSD’ye bağlı ON daha sıklıkla başlangıçta ciddi görme kaybı, iki taraflı tutulum ve kalıcı görme bozuklukları ile sonuçlanır.
Klasik olarak NMO yalnızca miyelit ve ON semptomlarını içeriyordu. Bununla birlikte, hastalığa neden olan antikorların keşfiyle birlikte, daha geniş bir hastalık belirtileri yelpazesi, NMOSD tanısında NMO ile birlikte gruplandırılmıştır. NMOSD, omurilik ve optik sinirden daha az sıklıkla beyin sapını etkileyebilir .
Beyin sapı veya üst servikal omurilikteki lezyonlar solunum yetmezliğine neden olabilir. Medulla oblongata’nın postrema bölgesindeki lezyonlar ağrı ve tonik spazmların yanı sıra kusma veya hıçkırıklara da neden olabilir .
Ek beyin lezyonları da yaygındır ancak sıklıkla asemptomatiktir (gerçi bilişsel eksiklikler ve depresyon , yetersiz teşhis edilen seküler olabilir ). Lezyonlar ayrıca çoğunlukla Aquaporin-4 – İmmünoglobulin-G (AQP4-IgG) NMOSD’de olmak üzere diensefalonu da etkileyebilir .
NMOSD , sinir sistemine otoimmün bir saldırıdan kaynaklanır . Vakaların %80’inden fazlasında aquaporin-4’e ( anti-AQP4+ ) karşı IgG otoantikorları nedendir ve geri kalan vakaların %10-40’ında MOG’ye karşı IgG antikorları nedendir. Geriye kalan vakaların nedeni hala bilinmiyor ve muhtemelen heterojendir.
Otoimmünitenin neden geliştiği büyük ölçüde bilinmemektedir. Çoklu genetik ve çevresel faktörlerin NMOSD gelişme riskini arttırdığı bilinmektedir. Özellikle AQP4-IgG pozitif NMOSD’de en güçlü risk faktörü kadın olmaktır. Çoklu insan lökosit antijeni (HLA) alelleri NMOSD ile ilişkilidir.
NMO geçmişte birçok sistemik hastalıkla ilişkilendiriliyordu. Bazı araştırmacılar diğer bazı vakaların paraneoplastik olabileceğine dikkat çekti . Görünüşe göre lupus, NMO-IgG otoantikorları üretebiliyor ve bu da lupus kaynaklı NMO vakalarına yol açıyor .
NMO-IgG’nin ( anti-AQP4 ) keşfi, nedenleri araştırmada yeni bir yol açtı .
Anti – AQP4 + çeşitleri
NMOSD’ye genellikle hücre zarında suyun zardan geçmesine izin veren bir kanal proteini olan aquaporin 4’ü (AQP4) hedef alan otoantikorlar neden olur . AQP4 monomerleri tetramerleri oluşturur ve tetramerler toplanır. AQP4, glifatik sistemin temeli olan astrositlerde bulunur . Bu nedenle, astrositler yarı seçici olarak yok edildiğinden, AQP4-IgG’yi içeren NMOSD, bir astrositopati veya otoimmün astrositik kanalopati olarak düşünülebilir .
Astrositler , kandaki maddelerin beyne girmesini önlemekten sorumlu bir sistem olan kan-beyin bariyerini (BBB) çevreler. Kandaki antikorların merkezi sinir sistemindeki (CNS) astrositlere ulaşması için, mekanizması tam olarak bilinmeyen BBB’yi geçmeleri gerekir. Bazı raporlar , BBB başarısızlığından sorumlu suçlular olarak metaloproteinaz -2 ve interlökin -6’ya işaret etmektedir . AQP4 /NMO- IgG’nin başlangıçta beyin omurilik sıvısına (CSP) erişimin olduğu postrema bölgesi gibi BBB eksikliği olan bölgeler yoluyla beyne girdiği konusunda geniş bir fikir birliği vardır . Her durumda,anti-AQP4 esas olarak intratekal olarak üretilir .
Astrositler içinde, AQP4 öncelikle kan damarlarına ve beyin zarlarına ( meninksler ) bitişik olan astrositik ayak çıkıntılarında bulunur . NMOSD beyin lezyonları, mikroskop altında görüldüğü gibi , kan damarları çevresinde IgG, İmmünoglobulin M (IgM), inflamatuar hücreler ve kompleman birikintilerini gösterir. AQP4-IgG , otoimmün yanıtta tamamlayıcı bir rol oynadığı düşünülen kompleman sisteminin bir aktivatörü olan IgG1 immünoglobulin ailesinin bir üyesidir . Astrosit kaybı ve bazen nöron ve oligodendrosit kaybı da vardır.. Astrositler dışındaki hücrelerin kaybı, ikincil inflamatuar hasarın veya astrosit fonksiyon bozukluğunun bir sonucudur.
NMOSD seçici olarak optik siniri, omuriliği ve beyin sapını etkiler. Bu seçicilik, bu yapılardaki artan AQP4 miktarıyla ve ayrıca optik sinir ve omurilikteki artan AQP4 agregat miktarıyla açıklanabilir. Postrema bölgesindeki artan BBB geçirgenliği, oradaki tutulumu açıklamaya yardımcı olur.
AQP4, merkezi sinir sistemi dışındaki dokularda (örneğin böbrekler) bulunur, ancak bu dokular, en azından kısmen merkezi sinir sistemi dışında otoimmün baskılayıcıların varlığı nedeniyle NMOSD’den etkilenmez.
NMOSD’de, geleneksel manyetik rezonans görüntülemede (MRI) normal görünen beyin dokusu alanları , difüzyon tensör görüntülemede (DTI) hasar gösterebilir , ancak bu, multipl sklerozla (MS) karşılaştırıldığında daha azdır .
NMO patolojisine yönelik araştırmaların çoğu omuriliğe odaklanmıştır . Hasar, inflamatuar demiyelinizasyondan beyaz ve gri maddelerin nekrotik hasarına kadar değişebilir . NMO’daki inflamatuar lezyonlar tip II lezyonlar ( kompleman aracılı demiyelinizasyon ) olarak sınıflandırılmıştır, ancak belirgin perivasküler dağılımları nedeniyle MS patern II lezyonlarından farklılık gösterirler . Bu nedenle inflamasyonun şekli çoğu zaman MS’te görülenden oldukça farklıdır.
AQP4-IgG seviyeleri, NMOSD hastalık aktivitesi ile kabaca ilişkilidir; bu seviyeler genellikle nüksetmeden önce artar ve remisyon sırasında düşer; daha yüksek seviyeler, daha şiddetli hastalık belirtileriyle ilişkilidir.
NMO- IgG -negatif vakalar daha az anlaşılmıştır. Bu vakalarda astrositlerin korunmuş olduğu görülüyor .
Anti – MOG + çeşitleri
NMO’da en sık görülen ikinci otoantikor, miyelin oligodendrosit glikoproteini (MOG) hedef alan MOG-IgG’dir. MOG, oligodendrositlerin yüzeyinde ve miyelin kılıflarının en dış yüzeyinde bulunan integral bir membran glikoproteinidir . İşlevi tam olarak bilinmemektedir. MOG sadece CNS’de mevcut olmasına rağmen (BBB ikisini ayırarak) MOG-IgG, merkezi sinir sistemi (CNS) dışında üretilir, bu da MOG’nin beyin omurilik sıvısı yoluyla lenf düğümlerine boşaltıldığı, otoimmün reaksiyon oluşumuna neden olduğu hipotezine yol açar.
MOG-IgG-pozitif NMOSD beyin lezyonları, mikroskobik olarak görüldüğü gibi, oligodendrositler ve aksonların korunmasıyla birlikte demiyelinizasyon, inflamatuar hücrelerin varlığı ve IgG ve kompleman birikintileri gösterir. [1] MOG-IgG düzeyleri kabaca hastalığın ciddiyeti ile ilişkilidir; düzeyler aktif hastalık sırasında daha yüksektir ve daha yüksek düzeyler daha şiddetli hastalık belirtileriyle ilişkilendirilir.
MS gibi benzer hastalıklarda MOG’a karşı antikorların çoğunlukla bulunmadığı düşünülmektedir. AQP4-IgG-negatif NMOSD içerisinde gruplandırıldığı söylenebilir .
Anti-AQP4 hastalığıyla birlikte anti-MOG hastalıkları NMO spektrumunun daha geniş bir bölümünü oluşturur. NMO vakaları, bu iki ana oto-antikordan herhangi birinin varlığına veya yokluğuna göre dört sınıfta sınıflandırılır .
Bu gruplar içinde sınıflandırılan çeşitli hastalıklar için klinik seyir ve tedaviye yanıt farklıdır; bu, NMO-Ab(-)/MOG-Ab(-) grubundakiler için daha iyi bir prognoz ve NMO’dakiler için daha kötü bir prognoz gösterir. -Ab(+)/MOG-Ab(+) grubu. MOG ile ilişkili NMO, konus tutulumu ile radyolojik olarak tanımlanabilir . Miyelin-oligodendrosit glikoprotein antikoru pozitif hastaların, spinal manyetik rezonans görüntülemede konu tutulumuna sahip olma olasılığı daha yüksekti .
Tanı
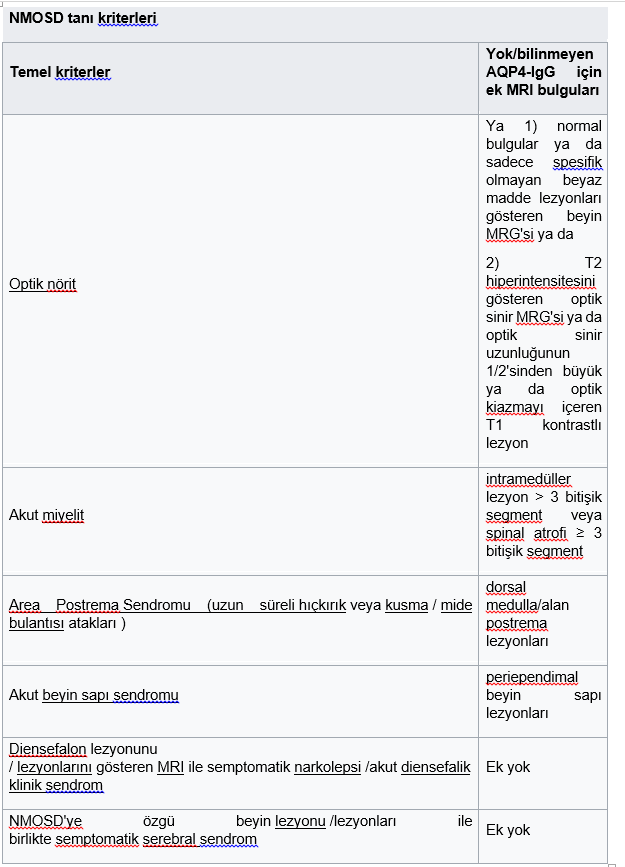
NMOSD tanısı , en son 2015’te birden fazla revizyona tabi tutulan konsensüs klinik kriterleri kullanılarak konur
Seropozitif AQP4 – IgG vakaları için tanı kriterleri, seronegatif AQP4-IgG vakalarına göre daha esnektir . AQP4-IgG tespit edilirse, NMOSD tanısı için alternatif tanıların dışlanmasıyla birlikte tek bir temel klinik kriter yeterlidir.
AQP4-IgG tespit edilmezse veya durumu bilinmiyorsa, NMOSD tanısı için her biri destekleyici MRI bulgularına sahip iki temel klinik kriterin yanı sıra alternatif tanıların dışlanması gerekir.
Nadiren, bazı anti-NMDAR tedavilerinin NMO ile tutarlı olduğu rapor edilmiştir . [30] Ön raporlar , nadir görülen NMO vakalarında diğer otoantikorların da rol oynayabileceğini düşündürmektedir .
MOG-IgG’li NMOSD, anti-MOG ile ilişkili ensefalomiyelitin bir belirtisi olarak kabul edilir . [33]
Spektrum Bileşenleri
NMO- IgG testinin geliştirilmesinden sonra NMO’yu kapsayan bozuklukların yelpazesi genişletildi. Spektrumun artık aşağıdakilerden oluştuğuna inanılıyor:
Yukarıda açıklanan tanı kriterlerine göre standart NMO
Uzunlamasına yaygın miyelit gibi tek veya tekrarlayan olaylar ve iki taraflı eş zamanlı veya tekrarlayan optik nörit gibi sınırlı NMO formları
Asya optik-spinal multipl skleroz (OSMS) veya AQP4 + OSMS. Bu varyant , MS gibi beyin lezyonlarını ortaya çıkarabilir ancak bazen optik-spinal MS olarak adlandırılan, merkezi sinir sistemi spektrumunun AQP4 negatif formu olan inflamatuar demiyelinizan hastalıklarla karıştırılmamalıdır.
Sistemik otoimmün hastalıkla ilişkili uzunlamasına yaygın miyelit veya optik nörit
Hipotalamus , periventriküler çekirdek ve beyin sapı gibi belirli beyin bölgelerindeki lezyonlarla ilişkili optik nörit veya miyelit
NMO -IgG negatif NMO: AQP4 antikoru – seronegatif NMO, teşhis açısından zorluk teşkil eder. Bazı vakalar anti- miyelin oligodendrosit glikoprotein (MOG) otoantikorlarıyla ilişkili olabilir .
Ayırıcı tanı
AQP4 -Ab-negatif NMO ayırıcı tanıda sorunlar yaratır . Oligoklonal bantların davranışı daha doğru bir tanı koymaya yardımcı olabilir. NMO’da oligoklonal bantlar nadirdir ve ataklardan sonra kaybolma eğilimindeyken, MS’te neredeyse her zaman mevcut ve kalıcıdırlar. MS’de nadir de olsa uzunlamasına lezyonların olabileceğinin ayırıcı tanıda dikkate alınması önemlidir .
Tanıda karşılaşılan diğer bir sorun ise MOG -ab düzeylerindeki AQP4-ab’nin tespit edilemeyecek kadar düşük olabilmesidir. Bazı ek biyobelirteçler önerilmiştir.
NMO, nadiren transvers miyelit olarak ortaya çıkan standart MS’e göre akut bir atak sonrasında genellikle daha ciddi sekellere sahip olması açısından MS’ten farklıdır . Ayrıca BOS’ta oligoklonal bantlar ve beyin MR’larında beyaz madde lezyonları NMO’da nadir görülür, ancak MS hastalarının %90’ından fazlasında görülür.
Son zamanlarda AQP4’ün varlığının standart MS’yi NMO’dan ayırdığı bulunmuştur; ancak MS heterojen bir durum olduğundan ve bazı MS vakalarının Kir4.1 kanalopatileri ( potasyum kanallarına karşı otoimmünite ) olduğu bildirildiğinden , NMO’yu MS spektrumunun bir parçası olarak düşünmek hâlâ mümkündür . Ayrıca, bazı NMO-AQP4(-) varyantları astrositopatik değil , demiyelinizandır .
NMO’da tümefaktif demiyelinizan lezyonlar olağan değildir, ancak yanlışlıkla interferon beta ile tedavi edilen birkaç vakada ortaya çıktıkları rapor edilmiştir .
Ayrıca Sjögren sendromuyla örtüşme de rapor edilmiştir.
AQP4 otoantikorunun keşfinden bu yana , NMO (tekrarlayan ve eş zamanlı optik sinir ve omurilik iltihabı ) tanısı için klinik gereklilikleri karşılamayan NMO benzeri semptomları olan hastalarda da ortaya çıktığı bulunmuştur.
Nöromiyelitis optika spektrum bozuklukları (NMOSD) terimi, AQP4 olmayan biyobelirteçlerle ilişkili vakaların dahil edilmesine izin verecek şekilde tasarlanmıştır . Bu nedenle, anti-AQP4’e bağlı tüm klinik varyantları ve ayrıca anti-MOG ile ilişkili ensefalomiyelit gibi diğer ilişkili olmayan ancak klinik olarak benzer sendromları içerir . MOG + ve AQP4+ antikorlarına sahip bazı vakalar bulunmuştur.
Bu varyantların standart NMO ile aynı tedavilere yanıt vermesi bekleniyor. Bazı yazarlar bu hastalıklar için ” otoimmün akuaporin-4 kanalopatisi ” adını kullanmayı önerirken ,diğerleri otoimmün olmayan kökenli AQP4 eksikliklerini de içeren daha genel bir terim olan “AQP4- astrositopati “yi tercih ederler .
Tedavi
NMO’nun tedavisi yoktur ancak tedavi edilebilir. Bazı hastalar iyileşir, ancak birçoğunda, bazı durumlarda ciddi olabilen görme ve uzuv bozuklukları kalır.
Uzun süreli nörolojik defisitlerin akut atakların kümülatif etkileri olması, akut tedavinin önemini vurgulamaktadır. [1] Geleneksel olarak ataklar, metilprednizolon IV ( Solu-Medrol ) gibi kısa süreli (3-5 gün) yüksek doz intravenöz kortikosteroidlerle tedavi edilir . Steroid tedavisine erken başlanmasının, akut ataklardan sonra görme ile ilgili sonuçları iyileştirdiği gösterilmiştir. Bununla birlikte, steroidlerin uzun vadeli sonuçları etkilediğine dair yüksek düzeyde kanıt yoktur; bu tedavi stratejisi benzer hastalıklara (idiyopatik optik nörit ve multipl skleroz) yönelik stratejiden ödünç alınmıştır.
Kortikosteroidlerin uygulanmasından sonra atakların ilerlemesi durumunda plazmaferez etkili bir tedavi olabilir . Bu tedavi, hastanın kendi kanının pompalanmasını, kan hücrelerinin plazmadan çıkarılıp bir solüsyonla karıştırılmasını ve ardından yeni kan karışımının tekrar pompalanmasını içerir.
NMO’nun nüksetmesini önlemek için genellikle profilaktik tedavi uygulanır; ancak böyle bir tedavinin kesin süresi tartışmalıdır.
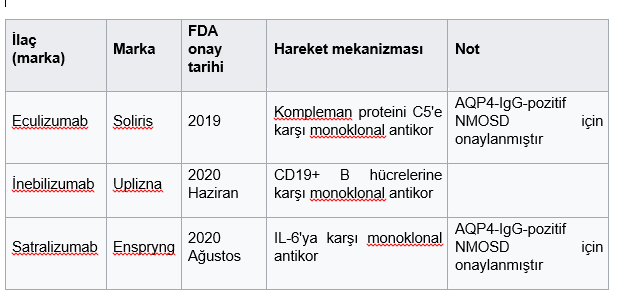
FDA onaylı farmasötikler
AQP4-IgG-pozitif NMOSD’ye karşı FDA onaylı, faz III klinik çalışmalarda etkili olduğu gösterilen ilaçlar ilk kez 2019’da kullanıma sunuldu. 2020 yılı itibarıyla dünya çapında en pahalı ilaçlar arasında yer alıyorlar. Hap formunda mevcut değiller, bu da yüksek fiyatlarının yanı sıra erişilebilirliklerini sınırlıyor. Bu yeni ilaçların AQP4-IgG-negatif NMOSD’ye karşı etkinliği bilinmemektedir.
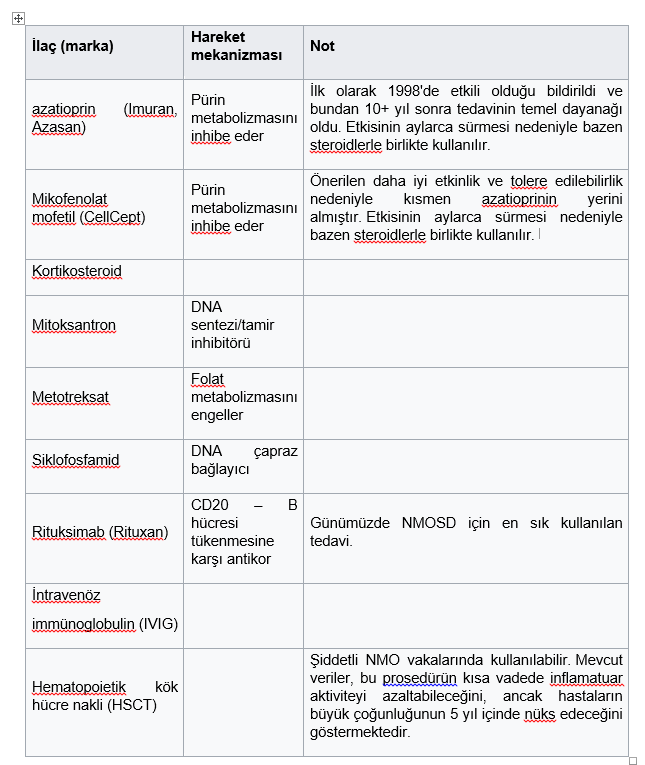
Endikasyon dışı tedaviler
Etkinliğini test eden faz III klinik araştırmaların olmamasına rağmen birçok tedavi kullanılmaktadır. Daha yeni, FDA onaylı ilaçlara göre ne üstünlüğü ne de üstünlüğü açıkça gösterilmiştir; nispeten ucuz olmaları ve hap formatında bulunabilmeleri de dikkate alındığında bu ilaçlar günümüzde standart tedavidir. Bu ilaçların çoğu bağışıklık sistemini çeşitli şekillerde etkiler.
MS tedavisinde kullanılan interferon-β , fingolimod , natalizumab ve alemtuzumab gibi bazı bağışıklık baskılayıcıların NMO hastalığının ilerlemesini kötüleştirdiğini ve NMO tedavisinde kullanılmaması gerektiğini unutmamak önemlidir .
Prognoz
Normalde birkaç hafta içinde bir miktar iyileşme görülür, ancak ciddi kalıntı semptomlar ve hatta sakatlık devam edebilir.
Hastalık monofazik olabilir, yani tek bir atak şeklinde olabilir ve daha sonra kalıcı remisyona uğrayabilir . Ancak hastaların en az %85’inde hastalığın tekrarlayan transvers miyelit ve/veya optik nörit ataklarıyla tekrarlayan bir formu vardır . Monofazik formdaki hastalarda transvers miyelit ve optik nörit aynı anda veya birkaç gün arayla ortaya çıkar. Öte yandan, tekrarlayan forma sahip hastaların ilk ataklar arasında haftalar veya aylar geçirme olasılığı daha yüksektir ve ilk transvers miyelit olayından sonra motor iyileşme daha iyi olur. Relapslar genellikle erken dönemde ortaya çıkar; hastaların yaklaşık %55’inde ilk yılda, %90’ında ise ilk beş yılda relaps görülür.
Tekrarlayan formun anti-AQP4+ seropozitif durumuyla, monofazik formun ise yokluğuyla ilişkili olması mümkündür . [65] MS’ten farklı olarak, NMO nadiren hastaların ataklar arasında remisyon olmadan artan nörolojik düşüşe sahip olduğu ikincil ilerleyici bir aşamaya sahiptir. Bunun yerine, akut ataklardan dolayı sakatlıklar ortaya çıkar.
Monofazik NMO hastalarının yaklaşık %20’sinde kalıcı görme kaybı , %30’unda ise bir veya her iki bacakta kalıcı felç vardır. Tekrarlayan NMO’lu hastaların %50’sinde beş yıl içinde körlük veya felç olur. Bazı hastalarda (bir çalışmada %33), servikal omurilikteki transvers miyelit, solunum yetmezliği ve ardından ölümle sonuçlandı . Ancak gelişmiş tanı kriterleri nedeniyle NMO’nun spektrumu genişledi; ve tedavi seçenekleri gelişti. Sonuç olarak araştırmacılar bu tahminlerin düşeceğine inanıyor.
AQP4 tutulumunun keşfedilmesinden bu yana , bazı araştırma çalışmaları anti-aquaporin 4 antikorlarını hedefleyen hedefe yönelik tedaviye odaklanmıştır . Antikorların uzaklaştırılmasında en köklü yöntem plazmaferezdir . Bir dizi ilaç üzerinde çalışılmaktadır: aquaporumab ( AQP4-IgG bağlanmasının patojenik olmayan antikor blokeri), sivelestat ( nötrofil elastaz inhibitörü) ve eculizumab ( kompleman inhibitörü).
Anti-AQP4 oto-antikorlarının birincil nedenlerine ilişkin çok az araştırma vardır. Bazı vakaların paraneoplastik olabileceği fark edilmiştir .
Ek olarak, AQP4’e karşı antikorlar dışında antikorlara sahip çeşitli NMO varyantları keşfedilmiş olup, bu da NMO’yu heterojen bir hastalığa dönüştürmektedir . NMO’da altı farklı hasar modeli rapor edilmiş olup, bu da altı farklı tipte oto-antikor olasılığını artırmaktadır . 2019 itibariyle bunlardan yalnızca üçü biliniyor. [
Yeni otoantikorlara yönelik araştırma
2016 yılında transvers miyelitte ( LETM ) ve atipik NMO’da bir otoantikor – glial fibriler asidik protein (GFAP) – bulundu ve bu durum otoimmün GFAP astrositopati kavramına yol açtı .
Araştırılan diğer otoantikor ise flotillindir . Seronegatif NMO ve bazı MS hastalarında bulunmuştur.
Son olarak, üzerinde çalışılan diğer proteinler connexin 43 ve anti- AQP1’dir , ancak 2015 yılı itibariyle bu proteinlerin katılımıyla ilgili yalnızca ilk raporlar mevcuttur .
AQP4 +/ MOG + grubu oldukça küçüktür ve aynı kişide tamamen ayrı iki hastalığın tesadüfü olarak değerlendirilebilir. Bu durumların doğrulanabileceği varsayılarak beş farklı NMO türü değerlendirilmektedir:
NMO bir otoimmün kanalopatiden ( AQP4 -Ab+) türetilmiştir – toplam NMO vakalarının yaklaşık %80’i
NMO, anti- MOG ile ilişkili ensefalomiyelitten türetilmiştir – toplam vakaların yaklaşık %10’u
Connexin-43 NMO
Aquaporin-1 ile ilişkili NMO , patern III MS ile ilişkili olabilir
Önceki antikorların yokluğuyla tanımlanan idiyopatik NMO
Antikor negatif nöromiyelitis optika
Bazı NMO vakaları otoantikorlara bağlı değildir . NMO ve MS arasında bir örtüşme oluştururlar.
2019 yılı itibarıyla yapılan bazı istatistiksel çalışmalar, antikor negatif NMO’nun üç gruba ayrılabileceğini ve bu sınıflandırmanın patojenik bir anlam taşıdığını göstermiştir.
Daha sonraki çalışmalar grup sayısını dörde kadar çıkardı.