Turner sendromunun doğumda saptanabilecek ilk klinik bulgusu aşağıdakilerden hangisidir?
A) Yele boyun
B) Kısa boy
C) Lokal lenfödem********************
D) Kubitus valgus
E) Madelung deformitesi
Turner sendromlu birçok hasta el ve ayak sırtında (lokalize) karakteristik lenfödem ve ensede gevşek cilt kıvrımları nedeni ile doğumda tanınabilir.
Çocuklukta yele boyun, düşük ense saç çizgisi, küçük mandibula, belirgin kulaklar, epikantal kıvrımlar, yüksek damak, ayrık meme uçları, geniş göğüs kafesi, kubitus vaigus ve hiperkonveks tırnaklar ile karşımıza çıkar.
Tanıdan çoğu kez seksüel olgunlaşmanın görülmediği pubertede kuşkulanılır. Kısa boy tüm Tumer sendromlu kızlarda kardinal (en sık) bulgudur. Büyüme yavaşlaması bebeklikte başlar ve giderek belirginleşir.
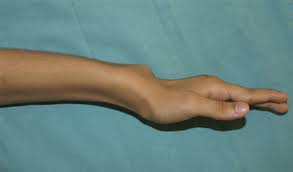
Madelung deformitesi Turner’de görülebilen, SHOX gen defektiyle ilgili, iskelet anormalliklerinden biridir.
Madelung deformitesi; karpal kemiklerin, radius ve protrude ulna arasında kama tarzında yerleşim gösterdiği bir el bileği deformitesidir. Distal radial büyüme plağının ulnar tarafının gecikmiş büyüme hızına ve eklem yüzeyinin rotasyonu sonucunda radiusun ulnar bölümünün rölatif kısalması ve eklem yüzeyinin rotasyonuna bağlı olarak oluştuğu düşünülmektedir. Radyolojik bulgular tanı koydurucudur. Posttravmatik, displastik, genetik ya da idiopatik olarak sınıflandırılabilir.
Turner sendromu ve Delesyon (Xp veya Xq)
Delesyon, bir kromozomun bir bölümünün eksik (silinmiş) olduğu anlamına gelir. Çok küçük bir kromozom parçası birçok farklı gen içerebilir ve delesyon, genetik materyalin kaybolmasına neden olur.
Turner Sendromuna sahip hastalarda Xp delesyon prevalansı yaklaşık %2′dir. Boy kısalığı, gonadal disgenezi ve karakteristik Turner Sendromu belirtileri özellikle kısa kolun tamamında delesyon gösteren hastalarda görülür. Ayrıca kısmi silmelerde fenotip değişkendir.
Xp22.33-Xp22.12 bölgesi, terminal bölgede bulunan SHOX genini içerir. Gen, X inaktivasyonundan kaçar ve işlevi doza bağlıdır.
Bu nedenle SHOX geninin haplo-yetersizliği büyüme geriliğine neden olur.
SHOX geni faringeal ark, uzuvlar, osteojenik hücreler, kemik iliği ve fibroblastlarda eksprese edilir. Genin haplo-yetersizliğinde boy kısalığının yanı sıra kısa metakarp, yüksek damak, kubitus valgus, Madelung deformitesi ve mezomelik displazi gibi iskelet anomalileri de görülebilir. SHOX genini veya onun düzenleyici bölgelerini içeren mutasyonlar, idiyopatik boy kısalığı olan hastaların yaklaşık %17’sinde, Leri-Weill Sendromlu hastaların ise %50-90’ında saptanabilmektedir.
Xp ve Yp üzerinde bulunan genler, kardiyovasküler sistemin normal gelişimi için gereklidir. Turner Sendromlu hastalarda konjenital kalp hastalıklarının doğum öncesi ve doğum sonrası ölümlerde önemli rolü vardır. En yaygın kardiyovasküler defektler hastaların %30’unda görülen biküspit aort kapağı ve aort koarktasyonudur. Ek olarak, genellikle ölümcül olan hipoplastik sol kalp sendromu hastaların yaklaşık %10’unda görülür. Özellikle, sol ventrikül çıkış yolu kusurları, Xp’nin terminal bölgesi ile ilişkilidir.
Sitogenetik çalışmalar, Xq13 ve Xq28 arasındaki bölgenin normal yumurtalık fonksiyonu için önemli olduğunu göstermiştir. Xq13-q21 bölgesi kritik bölge (CR) 1 olarak tanımlanır ve bu bölgenin proksimal delesyonları genellikle normal adet görme ve doğurganlık ile uyumludur. Xq23-q28’deki CR2’nin terminal ve interstisyel silmeleri, çoğunlukla erken yumurtalık yetmezliğinden sorumludur.
Yumurtalık fonksiyonunda önemli olan FMR1 geni, Xq27.3 lokusunda bulunur ve genin ekson 1 üçlü tekrarındaki genişlemeler, erken menopoz riskinin artmasıyla ilişkilidir.