Prenatal takipli 2 saatlik bebek oksipital bölge yerleşimli kitle nedeniyle yenidoğan yoğun bakım ünitesine yatırılıyor. Olgunun fizik muayenesinde yarık dudak-damak, mikrosefali ve polidaktili saptanıyor. Cinsiyet ayrımı yapılamıyor. Bu olgunun en olası tanısı aşağıdakilerden hangisidir?
A) Seckel sendromu
B) Fanconi sendromu
C) Meckel-Gruber sendromu
D) Edward sendromu
E) Smith-Lemli-Opitz sendromu
Cevap- C
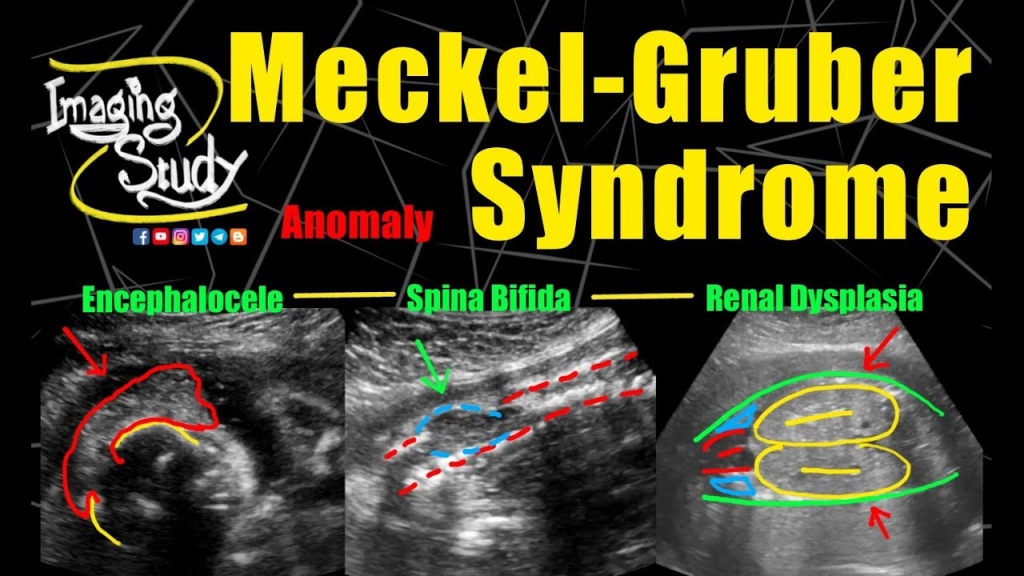
Bu olgunun en karakteristik özelliği oksipital bölgedeki kitledir. Oksipital bölgedeki kitle aksi ispat edilene kadar kraniyal meningosel veya kraniyal ensefaloseldir.
Kraniyal meningosel içi BOS dolu meningeal kese demektir. Kraniyal ensefalosel kesenin içinde serebral korteks, serebellum vea beyin sapı kısımlarının bulunmasıdır. En çok oksipital bölgeye yerleşir.

Oksipital ensefalosel denildiğinde aklımıza ilk gelmesi gereken sendrom Meckel-Gruber sendromudur. Olgularda ek olarak yarık dudak-damak, mikrosefali, anormal genitaller, konjenital nefroz ve polidaktili saptanabilir. Seçeneklerdeki tüm sendromların tek ortak karakteristiği hepsinin mikrosefali yapmasıdır.
Meckel-Gruber sendromu (Meckel sendromu), çok sayıda genin etkilenmesinden kaynaklanan, bu nedenle 15 fenotipi olan, otosomal resesif yolla aktarılan, siliyopatiler grubundan, kalıtsal bir sendromdur. Kistik böbrekler, beyin anomalileri ve ensefalosel, polidaktili ve karaciğer anomalileri başlıca bulgulardır. En sık görülen fenotip Meckel sendromu 1, öteki fenotiplerdeki bulguların çok büyük bir bölümünü içerir.
Bazı fenotipleri Joubert sendromuyla çakışan nitelikleri içermektedir.
Gruber sendromu , Disensefali splanchnocystica olarak da bilinir.
Oksipital ensefalosel
Bebeğin intrauterin gelişimi yetersizidir; doğum öncesi ve doğumdan kısa bir süre sonra kaybedilirler. Mikrosefali ve holoproensefali saptanır. Oksipital ensefalosel vardır. Boyun kısa ve kalın, alın bombesi yüksektir. Amnion sıvısı azlığı (oligohidroamnios) vardır; yüz yapısı ve üriner sistem bulguları Potter sequence’ını anımsatır. Göz(ler) küçüktür (mikroftalmi). Hipertelorizm ya da tersi (hipotelorizm) belirlenir. İris koloboması ile retina’nın yapısal bozuklukları (displazi) gözlerde saptanan önemli malformasyonlardır
Ağız büyük, ancak altçene küçüktür. Yarık dudak, yarık damak ve epiglot yarığı bulunur. Yenidoğanda dişlerin bulunduğu görülür (natal dişler). Dil lobülasyon nedeniyle engebelidir.
Dolaşım sisteminde, atrial ve ventriküler septal defektler ile aort koarktasyonu belirlenir. Akciğer tam gelişememiştir (pulmoner hipoplazi). Safra yolları displazileri ile dalak ve bağırsak anomalileri saptanır, anüs açıklığı olmayabilir (imperfore anüs).
Böbrek yokluğu (renal agenezis), polikistik böbrek, üriner kanallarda ve mesanede malformasyonlar saptanır. Adrenal bez küçüktür (hipoplazi). Dış genital organlarda hipoplazi ya da biçim bozuklukları izlenir. Uzun kemiklerde deformasyonlar ile parmak anomalileri (polidaktili, sindaktili, kıvrık parmaklar) vardır

Arnold-Chiari malformasyonları, Dandy-Walker malformasyonu, anensefali, beyin ve beyincik hipoplazisi, koku ve görme sistemlerinin gelişememesi ile corpus callosum yokluğu santral sinir sisteminde saptanan başlıca bulgulardır.
Göbek kordonunda bir adet atar damar (A.umblicalis) vardır.
Meckel-Gruber sendromu, renal kistik displazi , merkezi sinir sistemi malformasyonları (oksipital ensefalosel), polidaktili (postaksiyel), hepatik gelişimsel kusurlar ve oligohidramniyoza bağlı pulmoner hipoplazi ile karakterize, nadir, ölümcül siliyopatik bir genetik hastalıktır . [ kaynak belirtilmeli ] Meckel-Gruber sendromu, adını Johann Meckel ve Georg Gruber’den almıştır .
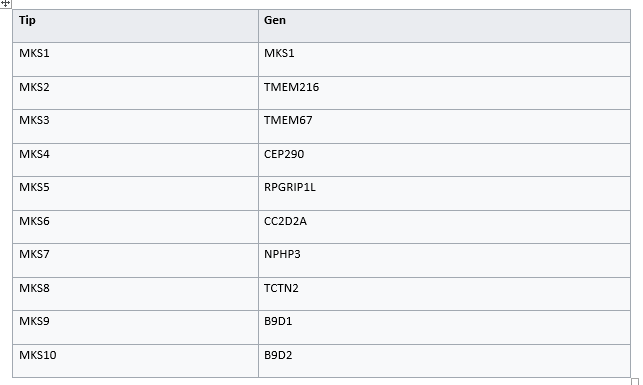
Meckel-Gruber sendromu (MKS), otozomal resesif geçişli ölümcül bir malformasyondur. Son zamanlarda iki MKS geni , MKS1 ve MKS3 tanımlanmıştır. Yakın zamanda yapılan bir çalışma , bu genler tarafından kodlanan yeni proteinler MKS1 ve meckelinin hücresel , hücre altı ve fonksiyonel karakterizasyonunu tanımladı
Bu ölümcül bozukluğun esas sorumlusu, bu protein üretiminin bozulmasıdır.
Genetik araştırmalardaki son bulgular , daha önce tıbbi literatürde ilişkili olarak tanımlanmayan çok sayıda genetik bozukluğun (hem genetik sendromlar hem de genetik hastalıklar ), aslında yaygın genetik hastalığın genetik temel nedeni ile yüksek oranda ilişkili olabileceğini öne sürdü. değişen, fenotipik olarak gözlenen bozukluklar . Bu nedenle Meckel-Gruber sendromu bir siliopatidir . Bilinen diğer siliopatiler arasında primer siliyer diskinezi , Bardet-Biedl sendromu , polikistik böbrek ve karaciğer hastalığı , nefronofitizis , Alström sendromu ve bazı retinal dejenerasyon formları bulunur .
MKS1 geninin siliopati ile ilişkili olduğu tespit edilmiştir
Displastik böbrekler, tanımlanan tüm vakaların %95’inden fazlasında yaygındır . Bu meydana geldiğinde, böbreğin içinde mikroskobik kistler gelişir ve yavaşça yok ederek orijinal boyutunun 10 ila 20 katına kadar büyümesine neden olur. Rahimdeki amniyotik sıvının seviyesi önemli ölçüde değişebilir veya normal kalabilir ve normal sıvı seviyesi tanının dışlanması için kriter olmamalıdır.
Oksipital ensefalosel tüm vakaların %60 ila %80’inde mevcuttur ve post-aksiyel polidaktili , tanımlanan toplam vaka sayısının %55 ila %75’inde mevcuttur. Uzuvların eğilmesi veya kısalması da yaygındır.
Normal karyotip varlığında klasik üçlünün üç fenotipik özelliğinden en az ikisinin bulunması tanıyı sağlamlaştırır. Düzenli ultrasonlar ve proaktif doğum öncesi bakım genellikle hamileliğin erken dönemlerinde semptomları tespit edebilir .