Geçirilmiş üst gastrointestinal sistem kanaması ve splenomegalisi olan 5 yaşındaki bir çocukta bakılan ultrasonografik incelemede her iki böbrekte multiple kistler görülüyor.
Bu hasta için en olası tanı aşağıdakilerden hangisidir?
- Konjenital hepatik fibrozis
- Portal ven trombozu
- Hepatorenal tirozinemi
- İdiyopatik portal hipertansiyon
- Alfa-1 antitripsin eksikliği
CEVAP: A
GIS kanama gibi portal hipertansiyon bulguları olan bir hastada kistik böbrek hastalığıda görülüyorda akla muhakkak kongenital hepatik fibrozis gelmelidir.
Konjenital hepatik fibrozis çocukluk çağında nadir görülen, otozomal resesif kalıtılan (bazende sporadik) görülebilen bir hastalıktır. Konjenital hepatik fibrozise eşlik etmesi en olası böbrek hastalığı polikistik böbrek hastalığıdır.
Konjenital Hepatik Fibrozis (KHF), karaciğerin safra yollarının anne karnındaki gelişimi sırasında (duktal plak malformasyonu) duraksaması sonucu oluşan, nadir görülen kalıtsal bir hastalıktır.
KHF’de karaciğer hücrelerinin (hepatositler) fonksiyonu genellikle korunur; ancak karaciğer içinde geniş lifli (fibrotik) bantlar oluşur. Bu durum, kanın karaciğerden geçişini zorlaştırarak portal hipertansiyona yol açar.
Klinik Belirtiler ve Bulgular
KHF dört farklı klinik tabloyla (veya bunların kombinasyonuyla) kendini gösterebilir:
- Portal Hipertansiyon (En Sık): Karaciğer fonksiyonları normal olmasına rağmen dalak büyümesi (splenomegali) ve yemek borusu varis kanamaları görülür.
- Kolanjitik Tip: Safra yollarındaki genişlemeler (Caroli hastalığı ile birlikte) nedeniyle tekrarlayan safra yolu enfeksiyonları ve ateş.
- Kolestatik Tip: Safra akışının bozulmasıyla gelişen sarılık.
- Latent (Sessiz) Tip: Erişkin yaşa kadar belirti vermeyebilir.
İlişkili Hastalıklar
KHF nadiren tek başına görülür; genellikle böbrek hastalıkları ile “siliofati” adı verilen bir grup genetik bozukluğun parçasıdır:
- Otozomal Resesif Polikistik Böbrek Hastalığı (ARPKD): En sık görülen birlikteliktir.
- Caroli Hastalığı: Karaciğer içi safra yollarının kistik genişlemesi. (KHF + Caroli = Caroli Sendromu).
- Joubert Sendromu veya Meckel-Gruber Sendromu gibi genetik durumlar.
Tanı Yöntemleri
Tanı koymak için multidisipliner bir yaklaşım gerekir:
- Laboratuvar: Karaciğer enzimleri (AST, ALT) genellikle normaldir ancak safra yolu tutulumu varsa GGT ve ALP yüksek olabilir.
- Görüntüleme: Ultrason (USG), BT veya MRCP ile karaciğerdeki fibrozis ve böbreklerdeki kistler incelenir.
- Altın Standart: Karaciğer biyopsisidir. Biyopsi, belirgin fibrozis ve safra kanallarındaki yapısal bozukluğu (duktal plak malformasyonu) gösterir.
- Genetik Testler: PKHD1 geni mutasyonlarının taranması.
Tedavi ve Yönetim
KHF’nin kesin bir tıbbi tedavisi yoktur; yönetim, komplikasyonları önlemeye yöneliktir:
- Varis Kanamaları: Endoskopik band ligasyonu veya portal basıncı düşüren ilaçlar.
- Şant Ameliyatları: İlaçla durdurulamayan kanamalarda kanı portal sistemden uzaklaştıran cerrahi işlemler (TIPS veya cerrahi şantlar).
- Enfeksiyon Kontrolü: Kolanjit atakları için antibiyotik tedavisi.
- Nakil: Karaciğer yetmezliği veya şiddetli kolanjit durumunda karaciğer nakli; böbrek tutulumu ağırsa karaciğer-böbrek nakli düşünülür.
Önemli Ayırıcı Tanı
KHF’yi sirozdan ayıran en kritik nokta, KHF’li hastaların karaciğer hücre fonksiyonlarının (albumin sentezi, pıhtılaşma faktörleri) uzun süre normal kalmasıdır. Yani çocukta varis kanaması var ama karaciğer testleri normalse KHF güçlü bir adaydır.
Konjenital Hepatik Fibrozis (KHF), çocukluk çağında görülen, portal fibrozis + korunmuş hepatosit fonksiyonu ile seyreden konjenital bir ductal plate malformasyonudur.
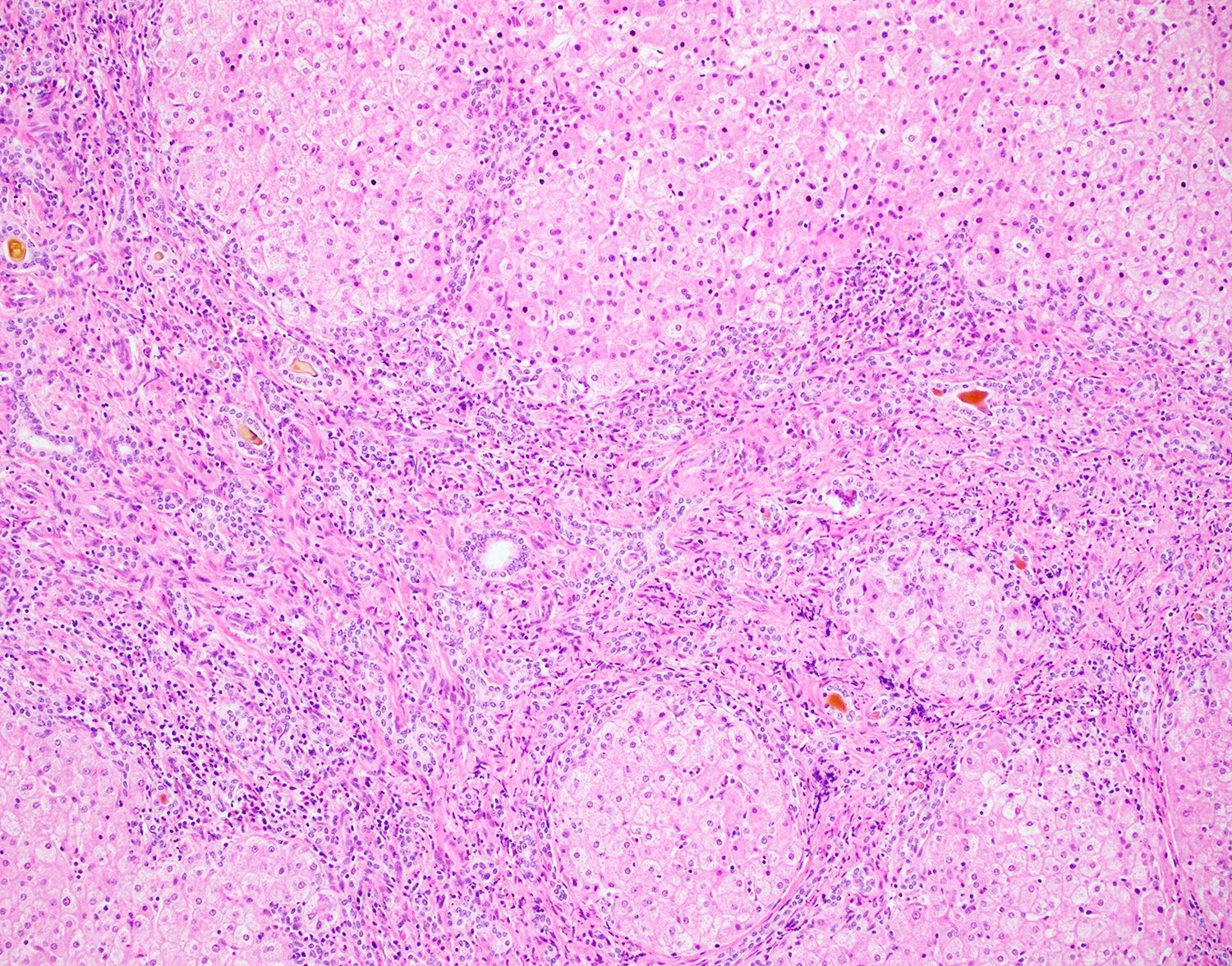


Otozomal resesif geçişlidir. İntrahepatik safra kanallarının embriyolojik gelişim bozukluğu (ductal plate malformasyonu) sonucu oluşur. Portal alanlarda yaygın fibrozis vardır. Hepatosit fonksiyonları genellikle korunmuştur
Bu nedenle Portal hipertansiyon var ama karaciğer yetmezliği yoktur
Normalde fetal dönemde ductal plate remodelize olur. KHF’de ise Bu remodelizasyon tamamlanamaz. Genişlemiş, anormal safra kanalları + portal fibrozis gelişir.
Klinik Bulgular; Genellikle çocukluk veya adolesan dönemde ortaya çıkar
1-Portal Hipertansiyon Bulguları olarak; Splenomegali, Trombositopeni (hipersplenizm), Özofagus varisleri ve Üst GİS kanaması ***
2-Karaciğer Fonksiyonlarında AST/ALT: normal veya hafif yüksek, Bilirubin: genellikle normal iken Albümin, PT: normaldir.
Portal HT var, hepatoselüler yetmezlik yok → ayırt edici
Eşlik eden durumlar en çok Böbrek anomalileri olup, sıklıkla Otozomal resesif polikistik böbrek hastalığı (ARPKD), Medüller sünger böbrek ve Nefronoftizidir.
KHF + ARPKD = klasik birliktelik
Tanıda Görüntüleme olarak; USG de hepatosplenomegali, Dopplerde portal HT bulguları ve MRCPde safra yolları değerlendirilir. Ama Kesin tanı Karaciğer biyopsisi ile konulur. Biyopside; Portal alanlarda fibrozis, Genişlemiş safra kanalları, Lobüler mimari korunmuştur
Ayırıcı Tanı
1-Siroz; lobüler mimari bozulur, karaciğer yetmezliği olur
2-Biliyer atrezi
3-Caroli hastalığı; KHF ile birlikte olabilir o zaman isimlendirmesi Caroli sendromu şeklinde olur.
Tedavi
Spesifik küratif tedavi yoktur. Tedavi hedefi; Portal hipertansiyonun komplikasyonlarının kontrolü, Varis profilaksisi ve tedavisi, Üst GİS kanamasında endoskopik girişimler ve İleri olguda Karaciğer ± böbrek nakli
Konjenital hepatik fibroziste portal hipertansiyon vardır ancak hepatoselüler fonksiyonlar genellikle korunmuştur; en sık ARPKD ile birliktedir.
KHF, ductal plate malformasyonuna bağlı gelişen, portal fibrozis ve portal hipertansiyonla seyreden ancak karaciğer yetmezliği yapmayan konjenital bir hastalıktır.