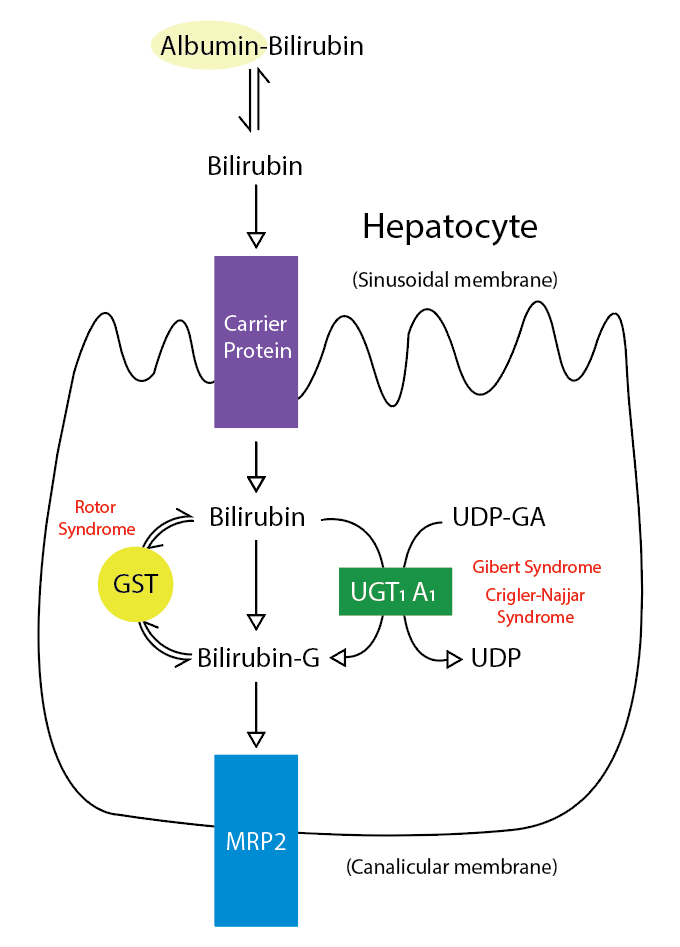
Yenidoğanın fizyolojik sarılığının hiperbilirubinemisi öncelikle konjuge olmayan bilirubine bağlıdır.
Gilbert sendomu orta derecede konjuge olmamış bilirubin yüksekliği ile karakterizedir
Crigler-Najjar sendromu ağır şeklinde (bazen tip I olarak da anılır) sıklıkla kernikterusa bağlı erken ölümle seyreder.
Dubin-Johnson sendromu hepatositlerde belirgin koyu renkli bir ***********pigment birikimi ve kanalikül membranından bilirubin taşınmasının bozukluğuna bağlı konjuge hiperbilirubinemi ile karakterizedir.
Rotor sendromu benzer şekilde safra taşınmasının bozukluğu ile oluşur fakat pigment birikimi görülmez.
Hereditary hyperbilirubinemia (bilirubin)
1-Unconjugated:
a-Gilbert’s syndrome
b-Crigler–Najjar syndrome
c-Lucey–Driscoll syndrome
2-Conjugated:
a-Dubin–Johnson syndrome
b-Rotor syndrome
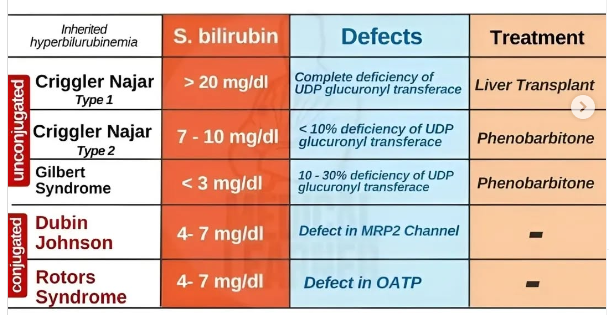
Gilbert sendromu , enzimin (UGT1A1) glukuronidasyon aktivitesinde %70-80 oranında azalma ile karakterizedir . UGT1A1 geni, insan kromozomu 2’de yer almaktadır .
Gilbert sendromu, Meulengracht sendromu, Gilbert-Lereboullet sendromu, Arias tipi hiperbilirubinemi, tip 1 hiperbilirubinemi, ailevi kolemi, ailevi hemolitik olmayan sarılık
Crigler–Najjar sendromu, kırmızı kan hücrelerindeki hemin parçalanmasıyla oluşan bir kimyasal olan bilirubinin metabolizmasını etkileyen nadir bir kalıtsal hastalıktır . Bu bozukluk , yüksek düzeyde konjuge olmayan bilirubinle sonuçlanan ve bebeklerde sıklıkla beyin hasarına yol açan bir tür hemolitik olmayan sarılıkla sonuçlanır. Bu bozukluk otozomal resesif bir şekilde kalıtılır . Yıllık görülme sıklığının 1.000.000’de 1 olduğu tahmin edilmektedir.
Bu sendrom, bazen Arias sendromu olarak adlandırılan I ve II tiplerine ayrılır. Bu iki tip, Gilbert sendromu , Dubin-Johnson sendromu ve Rotor sendromu ile birlikte, bilirubin metabolizmasındaki bilinen beş kalıtsal kusuru oluşturur . Gilbert sendromunun aksine, Crigler-Najjar sendromunun yalnızca birkaç vakası bilinmektedir.
Crigler-Najjar sendromunun belirtileri ve semptomları arasında sarılık, ishal, kusma, ateş, kafa karışıklığı, peltek konuşma, yutma zorluğu, yürüyüşte değişiklik, sendeleme, sık düşme ve nöbetler yer alır.
Bu, üridin difosfoglukuronat glukuronoziltransferaz ( UGT1A1 ) için kodlayan gendeki anormalliklerden kaynaklanır . UGT1A1 normalde hepatositler içinde bilirubin ve glukuronik asidin konjugasyonunu katalize eder. Konjuge bilirubin daha fazla suda çözünür ve safra ile atılır.
Lucey-Driscoll sendromu , bilirubin metabolizmasında rol oynayan enzimleri etkileyen otozomal resesif bir metabolik bozukluktur . Geçici ailevi neonatal konjuge olmayan hiperbilirubinemi olarak sınıflandırılan birkaç bozukluktan biridir .
Geçici ailevi neonatal hiperbilirubinemi olarak da bilinir.
Yaygın neden doğuştandır, ancak anne sütüyle yenidoğana geçen maternal steroidlerden de kaynaklanabilir . Emzirmeyle ilişkili sarılıktan farklıdır (emzirilen bebeklerin bilirubin seviyeleri mamayla beslenenlere göre daha yüksektir).
Crigler-Najjar sendromu ve Gilbert sendromuyla da bağlantılı olan UGT1A1 genindeki bir kusur , Lucey-Driscoll sendromunun konjenital formundan sorumludur
Dubin-Johnson sendromu; Konjuge hiperbilirubinemi, hepatositlerden safraya anyonik konjugatların kusurlu endojen ve ekzojen transferinin bir sonucudur. Bilirubin glukuronidlerinin bozulmuş safra atılımı, kanaliküler çoklu ilaç direnci proteini 2’deki (MRP2) bir mutasyondan kaynaklanır . Koyu pigmentli bir karaciğer, bilirubinden değil, polimerize epinefrin metabolitlerinden kaynaklanır.
Dubin-Johnson sendromu otozomal resesif kalıtım gösterir.
Dubin-Johnson sendromu, kromozom 10 üzerinde bulunan çoklu ilaç direnci proteini 2 genindeki (ABCC2) bir kusurdan kaynaklanır . Bu, otozomal resesif bir hastalıktır ve mutasyon sitoplazmik/bağlayıcı alanı etkilediğinden, muhtemelen bir fonksiyon kaybı mutasyonundan kaynaklanır.
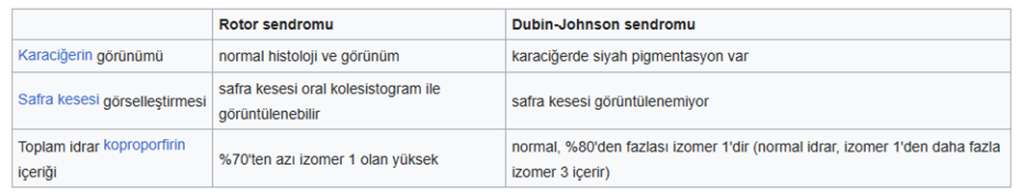
Rotor sendromu; bilirubin ve diğer bileşiklerin kandan karaciğere taşınmasından ve metabolize edilerek vücuttan atılmasından sorumlu iki proteindeki mutasyonlardan kaynaklanır.
SLCO1B1 ve SLCO1B3 genleri Rotor sendromunda rol oynar.
Safradaki önemli bir *** koproporfirin izomeri olan koproporfirin I , hepatositten dolaşıma geri taşınır ve idrarla atılır. Bu nedenle, Rotor sendromunda idrar koproporfirini yükselir.
Sülfobromoftalein (BSP) kullanılarak yapılan kolesintigrafi, boyanın *****safraya taşınma kapasitesinin %50’den daha az azaldığını ve ****hepatositlerdeki depolama kapasitesinin bu hastalıkta normal değerlere kıyasla 5 kattan fazla azaldığını göstermiştir.