Gorlin Sendromu; Nadir, otozomal dominant bir tablodur. Hayatın erken yaşlarında çok sayıda bazal hücreli karsinomla karakterizedir.
Beraberinde kemik, sinir sistemi göz ve genital sistem anomalileri içerir. Bu sendroma eşlik eden santral sinir sistemi tümörü medulloblastomdur.
Tümör süpresör gen olan PATCHED (PTCH) mutasyonu izlenir.
Gorlin Sendromu (OMIM #109400), bazal hücreli karsinomalar (BHK), iskelet anomalileri ve
çenede gözlenen çok sayıdaki kistlerle karakterize otozomal dominant kalıtımlı nadir bir hastalıktır.
Gorlin Sendromunun %50-85’inden PTCH1 genindeki mutasyonlar sorumludur
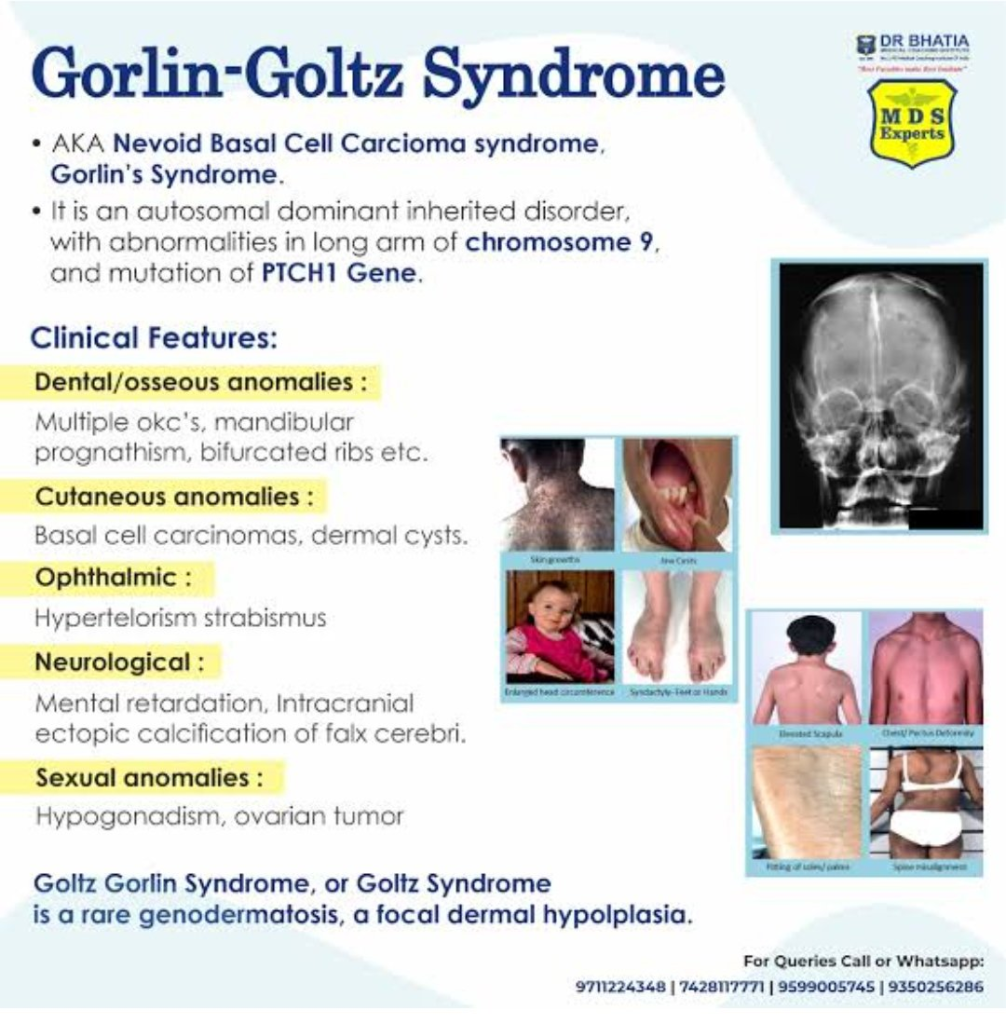
Gorlin sendromu (Basal cell nevi; Nevoid basal cell carcinoma; Gorlin-Goltz sendromu), otosomal dominant yolla aktarılan, deride tümörler ve kistler, çene kemiklerinde odontojen keratokistik tümörler, dudak ve damak yarığı, beyin zarında kireçlenmeler, avuçlarda ve tabanlarda çukurcuklar, çeşitli tümörler, mezanter kistleri ve kemik malformasyonları içeren kalıtsal bir sendromdur.
Frontal ve temporal kemikler makrosefali izlenimi verecek kadar geniş ve çıkıntılıdır (pagetoid görünüm). Beyinde, 20 yaş öncesi yapılan radyolojik incelemelerde, beyin zarlarının birleştiği yerlerde (falx cerebri) plaklar yapan kireçlenme alanları saptanır.
Yayvan bir burun sırtı vardır, paranazal sinüsler geniştir. Strabismus, konjunktiva kistleri, glokom ve iris defekti (koloboma) vardır.
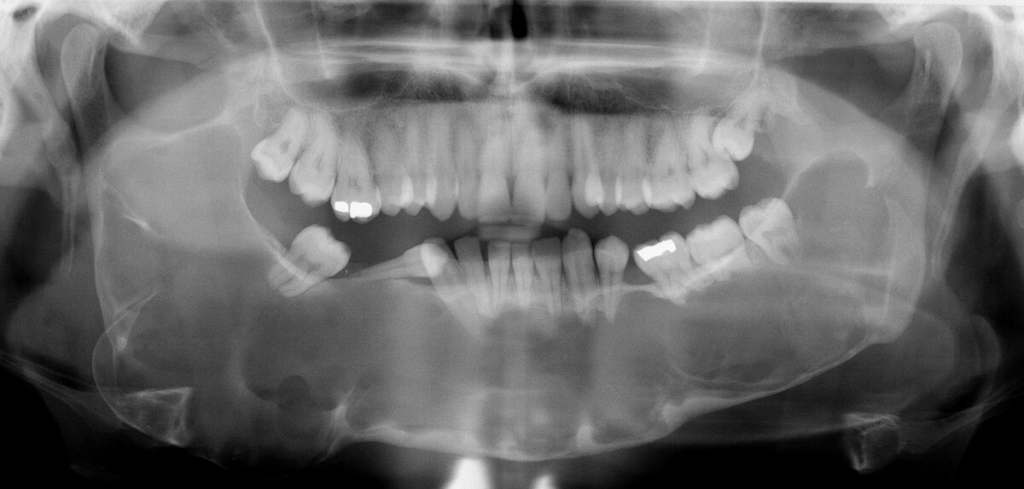
Çenelerde multipl keratokistik odontojen tümörler, yarık dudak ve yarık damak ile altçenede prognatizm saptanır.Altçenedeki kistik tümörler paresteziye neden olabilir.
Akciğerlerde kistik boşluklar olabilir. Kostalar yeterince gelişememiştir; yarık ya da kaynaşmış olabilirler. Bağırsaklar çevresinde mezanter kisti (lenfosel) gelişir. Vertebralarda kaynaşmalar, spina bifida, skolyoz ve kifoz (kifoskolyoz), kısa parmaklar iskelet sistemindeki önemli malformasyonlardır.
Deride multipl bazal hücreli karsinomlar vardır; tanı için 20 yaşından küçüklerde tek tümörün bulunması yeterlidir. Derialtı dokularda küçük kireçlenmelere rastlanır.
Tümör riski yüksektir. Ovaryum ve uterus fibromaları, ovaryum karsinomu, rabdomyoma, çenelerde ameloblastoma ve fibrosarkoma, çocuklarda medulloblastoma olası tümörlerin başlıcalarıdır.
Leiomyoma ve Wilms tümörü içeren olgular da bildirilmiştir.
Yukarıdakilere kıyasla daha seyrek saptanan bulgular da vardır; hipertelorizm, mikroftalmi, katarakt, retina anomalileri, kalp anomalileri, polidaktili, plevral kistler seyrek görülen bulgulardır.
Gorlin-Goltz sendromu oldukça nadir görülen otozomal baskın kalı-tımlı çoklu sistemik bir hastalıktır. Bazal hücreli karsinom, çene kistleri,frontal belirginleşme, vertebra anomalileri gibi iskelet anomalileri;palmoplantar çukurlanma ve falks serebri kalsifikasyonuyla belirgindir.Medulloblastom, fibrom, rabdomyom, leiomyosarkom vb. tümörlereeğilim vardır. Tanı büyük ve küçük klinik ve radyolojik ölçütleredayanmaktadır. Erken tanı ve tedavi, bu sendromun uzun dönemdesekellerini azaltmada büyük önem taşımaktadır.
Gorlin-Goltz sendromu veya Bazal hücreli nevus sendromu otozomal dominant olup ciltte görülebilen bazal hücreli karsinomalar, iskeletsel anomaliler ve çenelerde gözlenen çok sayıdaki kistle karakterizedir. Mandibular prognatizm, supraorbital sırtların belirginliği, frontal ve parietal şişkinlik, yüksek arklı damak, dudak ve damak yarığı, kaburgalarda çatallanma ile vertebra anomalileri bu sendromdaki gözlenebilen iskeletsel anomalilerdir. Odontojenik keratokistler bu sendromda karakteristiktir. Yaklaşık olarak hastaların %1i sadece çene lezyonlarına sahiptir. Lezyonlar sıklıkla mandibulada premolar-molar bölgesinde, farklı boyutlarda gözlenebilmektedir.
Gorlin Sendromu, Bazal Hücreli Nevus Sendromu ya da Gorlin Sendromu olarak da bilinen bazal hücreli karsinomalar (BHK), iskelet anomalileri ve çenede gözlenen çok sayıdaki kistlerle karakterize otozomal dominant kalıtımlı nadir bir hastalıktır. Hastalıkla ilişkili karakteristik kemik bulguları MÖ 2500-3000 yıllarından kalma iki Mısır iskeletinde gözlenmiştir
Sendrom ise 1960 yılında Dr. Gorlin ve Dr. Goltz tarafından bazal hücreli karsinom, çene kisti ve bifid kosta kliniği olan bir ailede tanımlanmıştır
Gorlin Sendromunun sıklığı toplumlar arası farklılık göstermektedir. Eski bir çalışmada 56000’de 1 sıklık bildirilirken daha güncel bir çalışmada 19000’de 1 kadar sık görülebildiği bildirilmiştir
Hastaların bir kısmının tanı almadığı ve asıl sıklığın çok daha yüksek olduğu düşünülmektedir. Hastalığın penetransı tam, ekspresivitesi ise değişkendir.
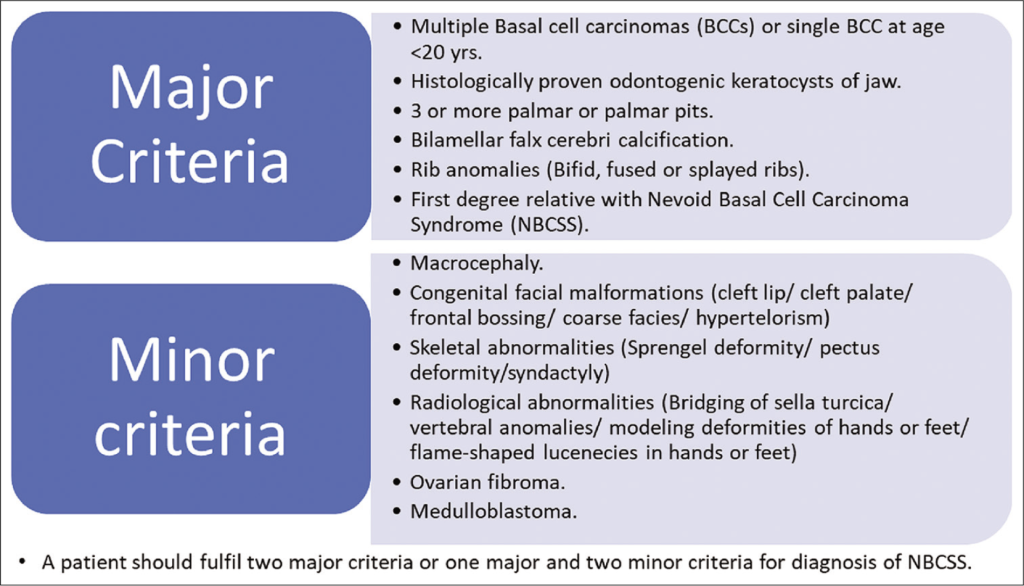
Hastalığın tanısı için bir major bulgu ve mutasyon saptanması; iki major bulgu veya bir major bulgu ve iki minör bulgu varlığı gerekmektedir.
Major bulgular:
1-İkiden fazla BHK ya da 20 yaş altında bir tane BHK öyküsünün olması
2-Çenenin odontojenik keratokistleri (histolojik olarak kanıtlanmış)
3-Üç ya da daha fazla palmoplantar çukurlanma
4- Falx cerebri kalsifikasyonu
5- Medulloblastom
6- Birinci derece akrabada Gorlin Sendromu tanısının olmasıdır.
Minör bulgular:
1- Makrosefali
2- Bifid ya da birleşik kosta
3- İskelet anomalileri ve radyolojik anomaliler
4- Lenfomezenterik kist
5- Over/kardiyak fibrom
6- Yarık dudak-damak
7- Göz anomalileridir
Gorlin Sendromunun bugüne kadar belirlenmiş üç geni mevcuttur. Bunlar PTCH1, PTCH2 ve SUFU genleridir
Klinik olarak kriterleri karşılayan hastaların %50-85’inde PTCH1 geninde, %5’inde ise SUFU geninde germline mutasyon saptanmaktadır
PTCH2 geni yeni saptanan bir gen olup mutasyonu oldukça nadirdir
PTCH1 geni “Drosophila patched” geni homoloğu olup 9q22.3 lokusunda bulunmaktadır. Bu gen 1447 amino asit uzunluğunda bir glikoprotein kodlamaktadır (6). Patched-1 proteini Sonic Hedgehog (SHH) yolağında transmembran reseptörü görevi görmektedir