
Çoklu endokrin neoplazi tip 2B (MEN 2B), ağız, gözler ve endokrin bezlerinde birden fazla tümöre neden olan genetik bir hastalıktır . Çoklu endokrin neoplazilerin en ciddi tipidir ve endokrin malignitelere ek olarak iyi huylu oral ve submukozal tümörlerin varlığıyla farklılaşır. İlk kez 1922’de Wagenmann tarafından tanımlanmış ve ilk kez 1965-1966’da ED Williams ve DJ Pollock tarafından bir sendrom olarak tanınmıştır.
RET genindeki p.Met918Thr patojenik varyantından kaynaklanır. Bu varyant medüller tiroid kanserine ve Feokromositoma’ya neden olabilir. Sunum, Marfanoid gövdesini, genişlemiş dudakları ve ganglionöromaları içerebilir.
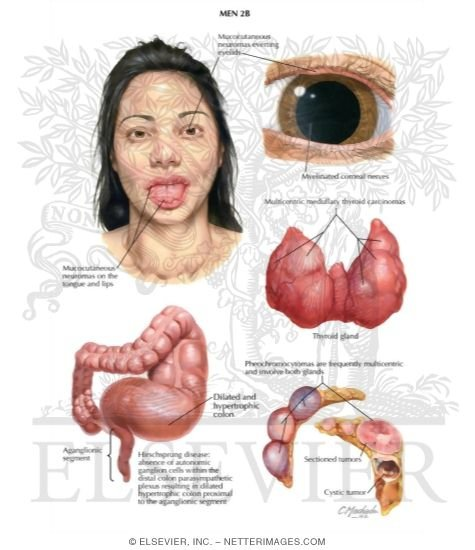
MEN 2B genellikle çocuk 10 yaşına gelmeden önce ortaya çıkar. Etkilenen bireyler genellikle uzun boylu ve ince yapılı, uzun yüzlü ve çıkıntılı, yağlı dudaklı olma eğilimindedir. İyi huylu tümörler ( neoplazmalar ), yaşamın ilk on yılında ağızda, gözlerde ve hemen hemen tüm organların submukozasında gelişir. [6] Medüller tiroid kanseri neredeyse her zaman, bazen de bebeklik döneminde ortaya çıkar. Çoğu zaman agresiftir. Adrenal bez kanseri ( feokromositoma ) vakaların %50’sinde görülür.

MEN 2B için Williams-Pollock sendromu, Gorlin-Vickers sendromu ve Wagenmann-Froboese sendromu gibi çeşitli eponimler önerilmiştir. Ancak hiçbiri sürekli kullanımı hak edecek kadar ilgi görmedi ve artık tıp literatüründe kullanılmıyorlar.
MEN2B’nin prevalansı iyi belirlenmemiştir ancak diğer epidemiyolojik değerlendirmelerden 600.000’de 1 ila 4.000.000’de 1 şeklinde elde edilmiştir.
Yıllık görülme sıklığının yılda 100 milyonda 4 olduğu tahmin edilmektedir.

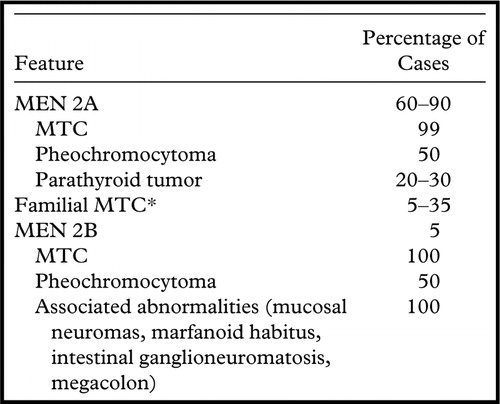
MEN2B’nin en yaygın klinik özellikleri şunlardır:
Uzun kemiklerin orantısız şekilde uzadığı uzun, ince, ” marfanoid ” vücut yapısı;
Ağız, dudaklar ve gözlerdeki mukozal yüzeylerin altındaki kitleler (aşağıda tartışılmıştır);
Bazen miyopati ile birlikte düşük kas kütlesi;
Gastrointestinal şikayetler, özellikle kabızlık;
Tiroidin medüller karsinomundan kaynaklanan semptomlar ;
Feokromasitoma kaynaklı semptomlar ;
Kraniosinostoz ;
Kuru gözler veya gözyaşı eksikliği;
Gecikmiş ergenlik.
Marfan sendromundan farklı olarak kardiyovasküler sistem ve göz merceği etkilenmez. [ kaynak belirtilmeli ] Mukozal nöromalar hastaların %100’ünde görülen en tutarlı ve ayırt edici özelliktir. Genellikle dudaklarda veya dilde çok sayıda sarımsı beyaz, sesil, ağrısız nodüller bulunur ve daha derin lezyonlar normal renktedir. Dudakların gövdesinde genişlemeye ve “balgamlı dudak” görünümüne neden olacak kadar nörom bulunabilir . Sklera ve göz kapaklarında da benzer nodüller görülebilir .
Histolojik olarak nöroma, omuriliğin arka kolonunu kaplayan hiperplastik , birbirine geçmiş Schwann hücreleri bantları ve miyelinli liflerden oluşan karakteristik bir adventif doku plağı içerir . Mukozal nöromalar, sıklıkla kalınlaşmış perinöryumlu , pleksiform bir düzende birbirleriyle iç içe geçmiş sinir hücrelerinden oluşur . Bu kıvrımlı sinir deseni, gevşek endonöryum benzeri fibröz stromanın arka planında görülür .
Nedenler
RET proto-onkogenindeki değişiklikler MEN2B’ye neden olur. Son yıllarda böyle bir varyasyona sahip olmayan hiçbir MEN2B vakası bildirilmemiştir. M918T varyantı tek başına vakaların yaklaşık %95’inden sorumludur. MEN2B’ye neden olan tüm DNA varyantlarının, ligandları dönüştürücü büyüme faktörü beta sinyal sisteminin bir parçası olan, hücre zarlarında bulunan bir reseptör molekülü olan RET proteini yoluyla sinyalleşmeyi arttırdığı düşünülmektedir . Vakaların yaklaşık yarısı otozomal dominant bir özellik olarak bir ebeveynden miras alınır . Diğer yarısı spontan mutasyonlar gibi görünmektedir, genellikle babaya ait alelden, [14] özellikle yaşlı babalardan kaynaklanmaktadır. De novo vakalarda cinsiyet oranı da eşitsizdir: erkek çocukların MEN 2B geliştirme olasılığı kız çocuklarına göre iki kat daha fazladır.
Teşhis
DNA testi artık MEN 2B tanısı koymak için tercih edilen yöntemdir ve neredeyse %100 duyarlı ve spesifik olduğu düşünülmektedir. Gordon ve ark. MEN2B’nin fiziksel fenotipine sahip, ancak RET geninde değişiklik olmayan ve malignite içermeyen farklı bir hastalık (“multipl mukozal nöroma sendromu”) vakalarını bildirdiler.
Bir kişide medüller tiroid karsinomu veya feokromasitoma olduğu tespit edildiğinde MEN2B bir tanı olarak kabul edilmelidir. DNA testi kullanıma sunulmadan önce, serum kalsitonin ölçümü MEN2B için en önemli laboratuvar testiydi. Kalsitonin, tiroidin “C” hücreleri tarafından üretilir; bu hücreler, MEN2B’de her zaman hiperplastik veya malign oldukları için normalden daha fazla kalsitonin üretirler. Kalsitonin düzeyleri, tiroidektomi sonrası medüller tiroid karsinomunun nüksetmesini saptamak için değerli bir belirteç olmaya devam etmektedir.
Luxol hızlı mavi boyama , bazı liflerin miyelin kılıfını tanımlar ve lezyonlu hücreler, S-100 proteini, kollajen tip IV, vimentin, NSE ve sinir filamentleri için immünohistokimyasal olarak reaksiyona girer. Daha olgun lezyonlar aynı zamanda EMA için de reaksiyon gösterecektir, bu da belirli miktarda perinöral farklılaşmaya işaret eder. Asit mukopolisakkaritlerden zengin erken lezyonlar alsiyan mavisi ile pozitif boyanır . Medüller tiroid kanseri mevcut olduğunda serum ve idrarda kalsitonin hormonu seviyeleri yükselir. Mikroskop altında, tümörler travmatik nöromaya çok benzeyebilir , ancak mukozal nöromanın akışlı fasikülleri genellikle daha tekdüzedir ve travmatik nöromanın iç içe geçmiş sinirleri, mukozal nöromanın kalın perinöryumundan yoksundur. Stromada inflamatuar hücreler görülmez ve nöral dokularda displazi yoktur.
Tedavi
Tedavi edilmezse MEN2B’li kişiler erken ölür. Resmi çalışmaların olmaması nedeniyle ayrıntılar eksiktir, ancak profilaktik tiroidektomi ve feokromasitoma sürveyansı yapılmadığı sürece genellikle 30’lu yaşlarda ölümün tipik olduğu varsayılmaktadır (aşağıya bakınız). Ancak aralık oldukça değişkendir: Erken çocukluk döneminde ölüm meydana gelebilir ve tedavi edilmeyen birkaç kişiye 50’li yaşlarında teşhis konulmuştur. Son zamanlarda hastalıkla ilgili daha geniş bir deneyim, “bireysel bir hastadaki prognozun önceden düşünülenden daha iyi olabileceğini düşündürmektedir.”
Tedavinin temelini tiroidektomi oluşturur ve tiroidde malignite saptanmasa bile MEN2B tanısı konulur konulmaz vakit geçirilmeden yapılmalıdır. Tiroidektomi olmadan, MEN2B’li hastaların neredeyse tamamında , MEN 2A’dan daha agresif bir formda medüller tiroid kanseri gelişir. [13] [19] Kanser 10 yaşından önce metastaz yapabileceğinden ameliyat için ideal yaş 4 yaş ve altıdır.
Vakaların %50’sinde adrenal bezlerin hormon salgılayan bir tümörü olan feokromositoma da mevcuttur. Etkilenen bireylerin tiroid ve adrenal kanser için yıllık tarama yaptırmaları teşvik edilmektedir.
Profilaktik tiroidektomi sağkalımı iyileştirdiğinden, MEN2B’li bir kişinin kan akrabaları, bozukluğun tipik belirti ve semptomları olmasa bile MEN2B açısından değerlendirilmelidir. Bu sendromun mukozal nöromaları asemptomatiktir ve kendi kendini sınırlar ve tedavi gerektiren herhangi bir sorun yaratmaz. Ancak estetik amaçlı veya sürekli travmaya maruz kalma durumunda cerrahi olarak çıkarılabilirler.