Sitrüllinemi , amonyak ve diğer toksik maddelerin kanda birikmesine neden olan otozomal resesif bir üre döngüsü bozukluğudur .
Her ikisi de farklı belirti ve semptomlara sahip olan ve farklı genlerdeki mutasyonlardan kaynaklanan iki sitrülinemi türü tanımlanmıştır. Sitrüllinemi, üre döngüsü bozuklukları adı verilen bir genetik hastalık sınıfına aittir . Üre döngüsü karaciğerde meydana gelen bir dizi kimyasal reaksiyondur . Bu reaksiyonlar , protein vücut tarafından enerji için kullanıldığında ortaya çıkan fazla nitrojeni işleyerek böbrekler tarafından atılan üreyi oluşturur .
Tip I sitrullinemi genellikle yaşamın ilk birkaç gününde belirginleşir. Etkilenen bebekler genellikle doğumda normal görünürler, ancak vücutta amonyak biriktiğinde enerji eksikliği ( uyuşukluk ), yetersiz beslenme, kusma , nöbetler ve bilinç kaybı gelişir. Bu tıbbi sorunlar çoğu durumda yaşamı tehdit edici olabilir. Tip I sitrülineminin daha hafif bir formu çocuklukta veya yetişkinlikte daha az görülür. Tip I sitrullinemiye neden olan gen mutasyonları olan bazı kişiler, bu bozukluğun belirti ve semptomlarını hiçbir zaman yaşamazlar. Sitrulinemi tip I tanısı kanda sitrülin seviyesinin yükselmesidir.
Tip I sitrülinemi, bozukluğun en yaygın şeklidir ve dünya çapında yaklaşık 57.000 doğumdan birini etkilemektedir. ASS genindeki mutasyonlar tip I sitrullinemiye neden olur. Bu gen tarafından üretilen argininosüksinat sentetaz enzimi , üre döngüsünün bir adımından sorumludur. ASS genindeki mutasyonlar enzimin aktivitesini azaltarak üre döngüsünü bozar ve vücudun nitrojeni etkili bir şekilde işlemesini engeller. Amonyak formundaki fazla nitrojen ve üre döngüsünün diğer yan ürünleri kan dolaşımında birikerek tip I sitrullineminin karakteristik özelliklerine yol açar.
Tip II sitrullineminin semptomları genellikle yetişkinlik döneminde ortaya çıkar ve esas olarak merkezi sinir sistemini etkiler . Karakteristik özellikler arasında kafa karışıklığı, anormal davranışlar (saldırganlık, sinirlilik ve hiperaktivite gibi), nöbetler ve koma yer alır. Tip II tanısı, sitrülin seviyesinin azalmasını ve amonyum iyonlarındaki artışın ölçülmesini içerir. Bu semptomlar hayatı tehdit edici olabilir ve bu tip kişilerde bazı ilaçlar, enfeksiyonlar ve alkol alımı tarafından tetiklendiği bilinmektedir .
Bebeklik döneminde neonatal kolestaz adı verilen karaciğer bozukluğu olan kişilerde tip II sitrülinemi de gelişebilir . Bu durum safra akışını engeller ve vücudun belirli besin maddelerini düzgün şekilde işlemesini engeller. Çoğu durumda semptomlar bir yıl içinde düzelir. Ancak yıllar hatta onyıllar sonra bu insanlardan bazıları yetişkin tip II sitrülineminin karakteristik özelliklerini geliştirir
Tip II sitrullinemi öncelikle Japon popülasyonunda bulunur ve tahminen 100.000 ila 230.000 kişide bir görülür. Tip II’nin Doğu Asya ve Orta Doğu popülasyonlarından insanlarda da görüldüğü rapor edilmiştir . SLC25A13 genindeki mutasyonlar tip II sitrülinemiden sorumludur. Bu gen , normalde belirli molekülleri mitokondrinin içine ve dışına taşıyan sitrin adı verilen bir protein yapar . Bu moleküller üre döngüsü için gereklidir ve ayrıca proteinlerin ve nükleotidlerin yapımında da rol oynarlar . SLC25A13’teki mutasyonlar tipik olarak üre döngüsünü engelleyen ve proteinlerin ve nükleotidlerin üretimini bozan herhangi bir fonksiyonel sitrin üretimini engeller. Ortaya çıkan amonyak ve diğer toksik maddelerin birikmesi, tip II sitrülinemi semptomlarına yol açar. Araştırmacılar, neonatal intrahepatik kolestazlı birçok bebeğin SLC25A13 geninde tip II sitrullinemili yetişkinlerle aynı mutasyonlara sahip olduğunu buldu.
Birden fazla tedavi yöntemi vardır. Düşük proteinli diyetler amonyak üretimini en aza indirmeyi amaçlamaktadır. Arginin, sodyum benzoat ve sodyum fenilasetat, amonyağın kandan uzaklaştırılmasına yardımcı olur. Kritik seviyelere ulaştığında amonyağın kandan uzaklaştırılması için diyaliz kullanılabilir. Bazı durumlarda karaciğer nakli başarılı olmuştur.
1-Citrullinemi tip I yada klasik sitrullinemi
a-Akut neonatal sitrülinemi tip 1 , yada sitrullinemi, akut neonatal, tip I; Klasik sitrülinemi tip 1
b-Yetişkin sitrülinemi tip 1 , yada sitrullinemi, yetişkin, tip I; Sitrüllinemi tip 1, geç başlangıçlı
2-Citrullinemi tip II ,yada sitrullinemi, yetişkin, tip 2; CTLN2; Sitrin eksikliği, yetişkin
Sitrin eksikliğine bağlı neonatal intrahepatik kolestaz
————————-
Üre siklus hastalıkları içinde hangisi transport defekti olarak bilinir?
- Hiperamonyemi Tip 1
- Hiperamonyemi Tip 2
- Sitrülinemi Tip 1
- Argininosüksinik asidüri
- Sitrülinemi Tip 2 **************
Sitrülinemi Tip 2, citrin ismi ile bilinenen ve aspartat ve glutamat transferinden sorumlu protein eksikliği ile karekterizedir.
Ayrıca Citrin “neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD)” eksikliğinde de azalır.
————–
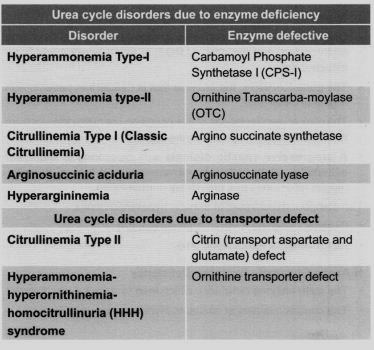
Üre döngüsü ile ilgili hastalıklar
1-Enzim defektleri
a-Hiperamonyemi Tip I; Karbomol fosfat sentetaz I eksikliği
b-Hiperamonyemi Tip II; Ornitin transkarba-moliyaz eksikliği
c-Sitrülinemi Tip I; Argininosüksinat sentetaz eksikliği
d-Argininosüksinik asidüri; Argininosüksinat liyaz eksikliği
e-Hiper argininemi; Arginaz eksikliği
2-Transport defektleri
a-Sitrülinemi tip II; Aspartat ve glutamat transportundan sorumlu sitrin defekti
b-Hiperamonyemi, Hiperornitinemi, Hipersitrülinemi (HHH) sendromu; Ornitin transporter defekti
——————————–
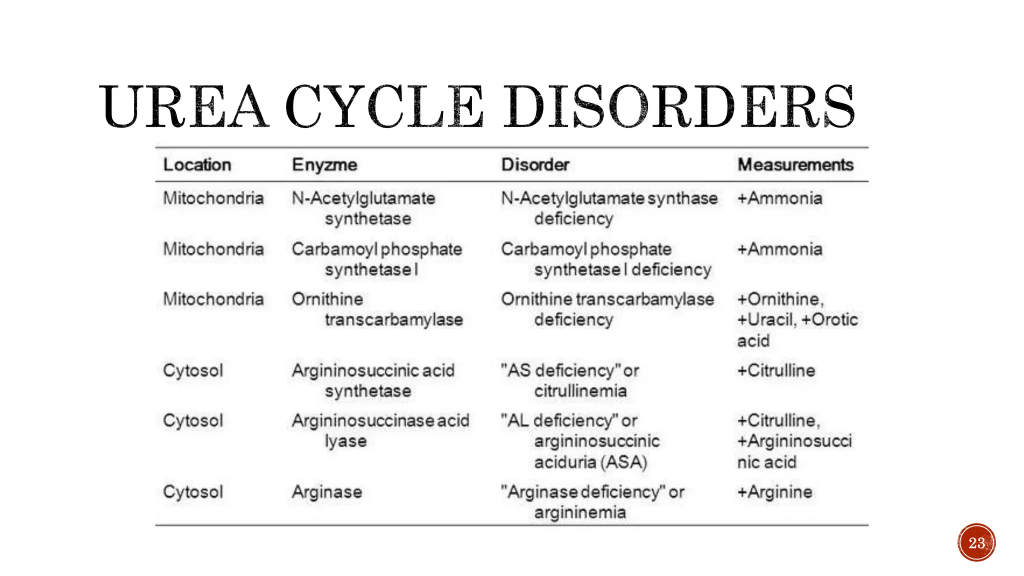
Üre döngüsü bozuklukları (UCD’ler), üre döngüsü yolunun altı enziminden (CPS1, OTC , ASS1, ASL , ARG1, NAGS) veya iki taşıyıcısından (ORNT1 veya sitrin) herhangi birindeki kalıtsal eksikliklerden kaynaklanır . Üre döngüsü kusurunun ciddiyeti, kusurlu proteinin yoldaki konumundan etkilenir. Şiddetli üre döngüsü bozukluğu olan bebeklerde doğumda semptom görülmez , ancak hızla serebral ödem , uyuşukluk, anoreksi, hiper veya hipoventilasyon, hipotermi, nöbetler , nörolojik duruş ve koma gelişir. Kısmi üre döngüsü enzim eksikliklerinde, ameliyat, uzun süreli açlık, tatil, peripartum dönem, daha yüksek protein alımını içeren basit beslenme değişiklikleri , tedavi (kortikosteroidler, valproat asit) gibi bir hastalık veya stres gibi yaşamın herhangi bir döneminde amonyak birikimi tetiklenebilir. plazma amonyak konsantrasyonunda çok sayıda hafif yükselmeye neden olur. Neredeyse her zaman baş ağrısı, konfüzyon, konvülsiyonlar, ataksi, ajitasyon ve gastrointestinal semptomların (mide bulantısı, kusma ve karın ağrısı) eşlik ettiği uyku bozuklukları, sanrılar, halüsinasyonlar ve psikoz ortaya çıkabilir.
Teşhis, plazma amonyak konsantrasyonuna ve kandaki amino asit anormalliklerine (sitrulin, ornitin ve arginin) dayanır . Moleküler genetik test, teşhis doğrulamanın birincil yöntemidir
En yaygın olanı X’e bağlı resesif bir durumdur, diğer eksiklikler ise otozomal resesif kalıtım modeline sahiptir.
———————-
Üre döngüsü bozukluklarından biri olan sitrülinemi, aminoasit metabolizmasında etkin argininosüksinat sentetaz enziminin eksikliğine bağlı, otozomal resesif geçişli bir hastalıktır. İnsidansı 1/100.000-1/230.000 arasında değişmektedir.
Yenidoğan dönemindeki ağır hastalık tablosundan, çocukluk çağı ve erişkinde ortaya çıkan çok hafif bulgulara dek farklı klinik tablolara neden olur. Başvuru sırasındaki klinik bulgular genellikle spesifik değildir ve neonatal metabolik dekompanzasyondan, erişkinlerdeki ensefalopati formlarına kadar farklı şekillerde karşımıza çıkabilmektedir.
Santral sinir sistemi hasarı ve ensefalopatinin oluşum mekanizması tam olarak anlaşılamamıştır. Yüksek serum amonyak konsantrasyonu ve astrositlerde glutamin birikiminin ensefalopatinin esas nedeni olduğu düşünülmektedir. İlk kez 1962 yılında Mc Murray ve ark. Tarafından tanımlanmıştır. Hastalık 9. kromozomdaki ASS1 ve SLC25A13 genlerinin mutasyonu sonucu ortaya çıkmaktadır.
Tip II sitrülinemi, otozomal resesif geçişli, arjininosüksinat sentetaz enzim eksikliğine bağlı gelişen erişkin başlangıçlı bir üre siklus defektidir. Bilinçte ani bozulma, huzursuzluk ve garip davranışlarla karakterize hiperamonyamik ensefelopati ataklarıyla giden kalıtsal bir metabolik hastalıktır. Görülme sıklığı Japonya’da 1/100.000 ile 1/230.000 arasında değişirken Amerika’da ve Avrupa’da bu oran daha düşüktür.
Argininosüksinat sentetaz enzimi eksiktir ve erişkin başlangıçlı form olarak bilinir. Kliniğe genellikle bilinçte bulanıklaşma, sersemlik, uykuya meyil, epileptik nöbet vb gibi hiperamonyeminin sonucu olan ensefelopati tablosu hakimdir. Ağır ensefelopati olmayan vakalarda semptomların diyet tedavisi ile gerilediği bilinmektedir.