Dilate kardiyomiyopatinin genetik belirleyicileri ve genotip-fenotip korelasyonları
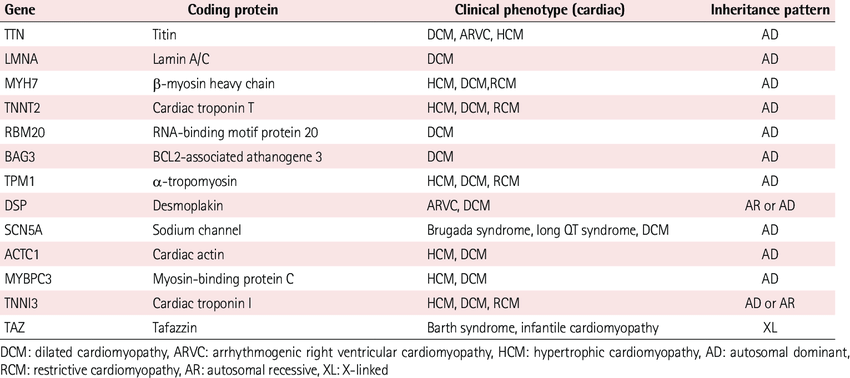
DCM’nin genetik formları vakaların neredeyse yarısını oluşturur ve şu ana kadar yaklaşık 40 nedensel gen tanımlandığı için derin genetik heterojenite ile karakterize edilir
Bu genler sarkomer, hücre iskeleti, nükleer zarf, sarkolemma, iyon kanalları ve hücreler arası bağlantılardaki çok çeşitli proteinleri kodlar. Bu genlerdeki spesifik mutasyonlar, çeşitli yolları ve hücresel yapıları değiştirerek, kas kasılma mekanizmasını, iyon kanallarının elektrolitlere duyarlılığını ve işleyişini, kalsiyum homeostazisini ve miyokarddaki mekanik kuvvetin oluşumu-iletimini olumsuz yönde etkiler. Bu genetik heterojenliğin ortak bir fenotiple sonuçlandığı gerçeği Bowles ve Towbin tarafından “Nihai Ortak Yol” hipoteziyle açıklanmıştır: farklı mutasyonlar, ortak bir yolda yer alan çeşitli proteinleri değiştirir ve bunların bozulması DCM’ye, hatta aritmojenik forma yol açar
DCM’nin pediatrik genetik formlarındaki ana kalıtım şekli otozomal resesiftir. Yetişkin popülasyonda, DCM’nin ailesel genetik formları vakaların %30-48’ini oluşturur, ana kalıtım paterni otozomal dominanttır (%56) ve genellikle eksik ve yaşa bağlı penetrasyon ve değişken ekspresyon ile karakterize edilirler. Klinik fenotip, başvuru yaşı, klinik özellikler ve ciddiyet bakımından yalnızca farklı aileler arasında değil, aynı ailenin üyeleri arasında da heterojendir. Hastalar, nakil gerektiren belirgin ilerleyici kalp yetmezliği gelişmeden önce birkaç yıl boyunca asemptomatik olabilir. Aritmiler, iletim sistemi bozuklukları ve ani ölüm sıklıkla hastalığın ilk belirtileridir
1-Sarkomerik genler
Sarkomer hem iskelet hem de kalp kasının temel kasılma birimidir. Sarkomerik proteinleri kodlayan genlerdeki mutasyonlar vakaların %5-10’unu oluşturur ve bazı vakalarda kuvvet üretimi ve iletimindeki kusurlarla ilişkilidir. Sarkomerik mutasyonlar DCM’nin önemli bir nedenidir, aynı zamanda prevalansın yaklaşık %60 olduğu hipertrofik kardiyomiyopatinin de önemli bir nedenidir. Bazen sarkomerik gen mutasyonları örtüşen fenotiplere yol açabilir.
DCM’de sarkomerik mutasyonların sarkomerik kasılma fonksiyonunu azalttığı (subklinik formlarda bile tespit edilen sistolik fonksiyon bozukluğu ile) hipotezi öne sürülmektedir; hipertrofik kardiyomiyopatide ise farklı sarkomerik mutasyonların fonksiyon mekanizmalarının kazanılması yoluyla kuvvet oluşumunu arttırdığına inanılmaktadır
a-Miyozin proteinlerini ( MYH6, MYH7 ve MYBPC3 )
b-Aktin proteinlerini ( ACTC1 ve ACTC2 ) ve tropomiyosin proteinini ( TPM1 ) kodlayan genlerdeki mutasyonlar , aktinin miyozine/miyozinden doğru şekilde bağlanması-ayrılmasında değişikliklerle sonuçlanır
c-Titin (TTN); Son çalışmalar , protein kesen mutasyonların ailesel DCM’nin %25’inde ve idiyopatik DCM’nin %18’inde tespit edilmesi nedeniyle DCM’de en sık yer alan sarkomerik genin titin ( TTN ) olduğunu göstermiştir. Titin, kalpte yüksek düzeyde eksprese edilen ve sarkomerin Z çizgisinden M çizgisine kadar uzanan, bilinen en büyük insan proteinidir. TTN sırasıyla diyastolik ve sistolik fonksiyonu korumak için hem pasif kuvvet hem de esneklik sağlar. Ayrıca sarkomerin dizilişini ve uzunluğunu da düzenler. DCM’de kesme mutasyonlarının rolü kabul edilirken, yanlış anlamlı varyantların patojenik ve prognostik rolü hala tartışılmakta ve araştırılmaktadır.
Sarkomer varyantlarının etkisine ilişkin boylamsal prognostik verilerin sınırlı olmasına rağmen, Merlo ve ark. sarkomerik nadir varyant taşıyıcılarının, özellikle 50 yaşından sonra, taşıyıcı olmayanlara kıyasla ölüme veya kalp nakline doğru daha hızlı bir ilerleme gösterdiğini gözlemledi
2-Nükleer proteinler
Nükleer proteinler lamin A ve C, nükleer zarfın laminasını oluşturan ara filamentlerdir. Bunlar, kromozom 1 üzerindeki aynı LMNA gen haritalaması tarafından kodlanan iki izoformdur. Bu proteinler, çekirdekte yapısal/mekanik işlevlere sahiptir ve DNA’nın replikasyonunu ve transkripsiyonunu düzenler
a-LMNA mutasyonları, aritmiler ve iletim hastalığı olan DCM, Limb-Girdle Musküler Distrofi, Emery-Dreifuss Musküler Distrofi ve otozomal dominant kısmi lipodistrofi dahil olmak üzere çeşitli fenotiplerle ilişkilidir. DCM hastaları arasında LMNA mutasyonlarının rapor edilen prevalansı, otozomal dominant kalıtım modeliyle yaklaşık %8’dir
Klinik açıdan bakıldığında, LMNA mutasyonu taşıyan DCM hastalarında hastalık erken başlangıçlıdır, kardiyak iletim bozuklukları, yüksek kreatinin kinaz düzeyleriyle birlikte iskelet kası tutulumu vardır ve hayatı tehdit eden veya malign ventriküler aritmiler açısından yüksek risk altındadırlar. ani ölüm
LMNA ile etkileşime giren nükleer zarfın diğer proteinleri, bir DCM fenotipine neden olabilir.
b-Timopoietin, kromozom 12 üzerindeki TMPO ( LAP2 ) geni tarafından kodlanan Lamin A/C ile etkileşime giren bir proteindir : LAP2’deki bir mutasyon, yaklaşık %1’lik düşük bir prevalansla DCM ile ilişkilendirilmiştir
3-İyon Kanalları proteinleri
DCM’de yer alan iyon kanalları proteinleri arasında sodyum kanalı ( SCN5A ) ve fosfolamban ( PLN ) mutasyonları açıklanmıştır ( 36 ) ( 37 )
a- Sodyum Kanalı ( SCN5A )
b- Fosfolamban ( PLN )
SCN5A mutasyonları, DCM ailelerinin %1,7’sini oluşturur ve hastalığın erken başlangıcı, atriyal fibrilasyon veya ventriküler taşikardi gibi aritmiler, sinüs düğümü disfonksiyonu ve iletim hastalığı ile karakterize edilen aritmojenik bir fenotip ile ilişkilidir
SCN5A mutasyonları aynı zamanda Long QT Sendromu ve Brugada Sendromu gibi diğer aritmik bozukluklarla da ilişkilidir.
PLN mutasyonlarının kalsiyum pompasının (SERCA2a) inhibisyonu yoluyla DCM’ye yol açtığı düşünülmektedir.
Ayrıca, PLN genindeki (R14del) spesifik bir mutasyonun, malign ventriküler aritmiler ve geç ergenlikten itibaren son dönem kalp yetmezliği açısından yüksek riskle ilişkili olduğu ve DCM fenotipine veya aritmojenik sağ ventriküler kardiyomiyopatiye neden olabileceği rapor edilmiştir ( 40 ).
4-Hücre iskeleti proteinleri
a-Desmin ( DES ) mutasyonları, iskelet miyopatisi, karışık iskelet-kardiyak hastalığı (“desmin ile ilişkili miyopati”) ve kardiyomiyopati (DCM’nin yanı sıra hipertrofik veya kısıtlayıcı kardiyomiyopatiler) dahil olmak üzere bir dizi fenotipe neden olabilir. DCM’den önce tipik olarak iskelet miyopatisi gelir ve iletim kusurlarıyla ilişkilendirilebilir. DCM’de DES mutasyonlarının bildirilen prevalansı DCM vakalarının yaklaşık %1-2’sidir
b-Cypher/ZASP ( LDB3 ) mutasyonları, Vatta ve arkadaşları tarafından 2003 yılında tanımlanmıştır ve iskelet tutulumu olsun veya olmasın, saf DCM veya sol ventriküler sıkışmasız DCM ile ilişkili olduğu düşünülmektedir ve pediatrik popülasyonda daha yaygın olarak bulunmaktadır
c-Distrofini ( DMD ) kodlayan gendeki mutasyonlar Duchenne kas distrofisine neden olur; daha hafif, daha sonra başlayan Becker kas distrofisine ve kas tutulumunun olmadığı veya subklinik olduğu X’e bağlı DCM’ye neden olur
DMD genleri X kromozomu üzerinde haritalanır, bu nedenle kalıtım modeli X’e bağlıdır ( 45 ) . Distrofin, distrofin glikoprotein kompleksi ile birlikte hücre iskeleti ile hücre dışı matris arasında bir bağlantı görevi görür ve bu nedenle mutasyonları, kuvvet aktarımının değişmesine ve ilerleyici hücre ölümüne neden olur. Supraventriküler aritmiler, atriyoventriküler bloklar ve sağ dal bloğu ile birlikte DCM, X’e bağlı DCM’yi karakterize eder ve Duchenne kas distrofisinde geç bir komplikasyon olarak ortaya çıkarken, Becker kas distrofisinde daha az sıklıkta görülür.
5-Diğer genler ve proteinler
Bir dizi başka gen ve mekanizma DCM fenotipine yol açabilir
DCM’nin genetik temeline ilişkin en son keşifler arasında, çizgili kaslardaki Z diskinde lokalize olan, anti-apoptotik fonksiyona sahip bir ko-şaperon proteini olan BCL2 ile ilişkili atanojen 3’ü kodlayan BAG3 geni yer almaktadır.
a-BAG3 mutasyonlarının tahmini prevalansı Japon ailesel DCM hastalarının %2,8’idir. Z-disk düzeneğine müdahale ederek ve metabolik stres altında apoptotik hücre ölümünü indükleyerek DCM’ye neden olduğu düşünülmektedir Ancak bu gendeki mutasyonların neden olduğu fonksiyonel anormallikler tam olarak anlaşılamamıştır.
b-Lamin alfa-4 ( LAMA4 )
c-Fukutin ( FKT ) gibi hücre dışı matriks proteinleri tanımlanmış ve DCM ile ilişkilendirilmiştir: sırasıyla sinyal yollarını bozarak ve hücre yüzeyi moleküllerini değiştirerek DCM fenotipine yol açabilirler