Progresif ailesel intrahepatik kolestaz (PFIC), safra epitelyal taşıyıcılarındaki kusurların neden olduğu bir grup ailesel kolestatik durumdur . Klinik tablo genellikle ilk olarak çocukluk çağında ilerleyici kolestaz ile ortaya çıkar . Bu genellikle gelişme geriliğine , siroza ve karaciğer nakli ihtiyacına yol açar .
Progresif ailesel intrahepatik kolestaz türleri aşağıdaki gibidir :
Tip 1, Byler hastalığı olarak da bilinir
Tip 2, ABCB11 eksikliği veya BSEP eksikliği olarak da adlandırılır
Tip 3, ABCB4 eksikliği veya MDR3 eksikliği olarak da adlandırılır
Tip 4, TJP2’deki mutasyondan
Hastalığın başlangıcı genellikle 2 yaşından öncedir, ancak hastalara ergenlik döneminde bile PFIC tanısı konulmuştur . Üç antiteden PFIC-1 genellikle en erken ortaya çıkar. Hastalar genellikle erken çocukluk döneminde kolestaz , sarılık ve gelişme geriliği ile başvururlar . Yoğun kaşıntı karakteristiktir; ergenlik döneminde başvuran hastalarda intiharla ilişkilendirilmiştir . Hastalarda yağ malabsorbsiyonu olabilir , bu da yağda çözünen vitamin eksikliğine ve osteopeni dahil komplikasyonlara neden olabilir .
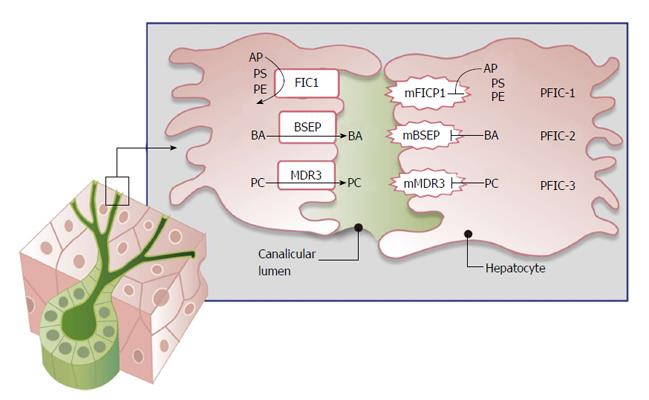
Progresif ailesel intrahepatik kolestaz otozomal resesif bir şekilde kalıtsaldır .
PFIC-1’e , membranlar boyunca fosfolipid translokasyonundan sorumlu olan P tipi ATPase proteini FIC- 1’i kodlayan bir gen olan ATP8B1’deki çeşitli mutasyonlar neden olur. Daha önce Byler hastalığı ve Grönland-Eskimo ailesel kolestazı olarak bilinen klinik antiteler olarak tanımlanmıştı . PFIC-1’li hastalarda , FIC-1’in bağırsakta ekspresyonuna bağlı olarak aşağıdaki klinik özelliklere ek olarak sulu ishal de görülebilir . ATP8B1 mutasyonunun kolestaza nasıl yol açtığı henüz tam olarak anlaşılamamıştır.
PFIC-2’ye , safra tuzu ihracat pompasını veya BSEP’i kodlayan gen olan ABCB11’deki çeşitli mutasyonlar neden olur. BSEP’yi eksprese eden tek hücre tipi olan hepatositlerde safra tuzlarının tutulması, hepatoselüler hasara ve kolestaza neden olur.
PFIC-3’e , fosfatidilkolin translokasyonundan sorumlu bir floppazı kodlayan çoklu ilaç direnç proteini 3’ü (MDR3) kodlayan gen olan ABCB4’teki çeşitli mutasyonlar neden olur . Kusurlu fosfatidilkolin translokasyonu safrada fosfatidilkolin eksikliğine yol açar. Fosfatidilkolin normalde safra asitlerine eşlik ederek safra epitelinin zarar görmesini önler. MDR3 eksikliği olan hastaların safrasındaki serbest veya “şaperonsuz” safra asitleri kolanjite neden olur . PFIC-3 belirgin derecede yüksek bir GGT ile ilişkili olduğundan biyokimyasal olarak bu dikkate değerdir.
Bugüne kadar tanımlanan üç PFIC formunun kalıtım modeli otozomal resesiftir .
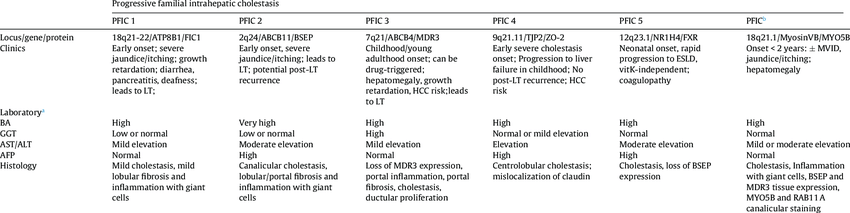
Karaciğer biyopsileri tipik olarak kolestaz (safra tıkaçları ve safra enfarktüsleri dahil), kanal hipoplazisi, hepatoselüler hasar ve Bölge 3 fibrozis kanıtlarını gösterir . Dev hücre değişimi ve hepatoselüler hasarın diğer özellikleri PFIC-2’de PFIC-1 veya PFIC-3’e göre daha belirgindir. Bugüne kadar tanımlanan tüm PFIC formlarındaki son dönem hastalık, peri-portal bölgelerde kanal proliferasyonu ile fibrozis arasında köprü kurulmasıyla karakterize edilir. [6]
Biyokimyasal belirteçler, PFIC-1 ve -2 için normal bir GGT’yi ve PFIC-3 için belirgin şekilde yüksek bir GGT’yi içerir. Serum safra asidi seviyeleri ciddi oranda yükselmiştir. Patoloji safra hücrelerindeki anatomik bir problemin aksine bir taşıyıcıdan kaynaklandığından , genellikle kolestazda görüldüğü gibi serum kolesterol seviyeleri tipik olarak yükselmez .
Alkalen fosfataz (ALP) sevinde artışa gamaglutamil transferaz (GGT) artışının eşlik ettiği durum aşağıdakilerden hangisidir?
A) Ursodeoksikolik asit kullanimi
B) Benign rekurren intrahepatik kolestaz I
C) Progresif familyal intrahepatik kolestaz II
D) Progresif familyal intrahepatik kolestaz III *****
E) Gebeligin intrahepatik kolestazi
Kolestaz durumlarında alkalen fosfataz (ALP) sevinde artışa gamaglutamil transferaz (GGT) seviyesinde artış eşlik eder. Ursodeoksikolik asit (UDCA) tedavisi kullanımı, benign rekurren intrahepatik kolestaz (BRIK) I ve II, progresif familyal intrahepatik kolestaz (PFIK) I ve II ve gebeliğin intrahepatik kolestazında ALP artışına GGT artışı eşlik etmemektedir. Progresif familyal intrahepatik kolestaz III de ise GGT artışı görülmektedir
Tedavi
İlk tedavi, kolestaz ve kaşıntıyı tedavi etmek için aşağıdakileri içeren ajanların kullanımını içeren destekleyicidir:
Ursodeoksikolik asit
Kolestiramin
Rifampin
Dirençli vakalarda nalokson
Kısmi dış biliyer saptırma (PEBD) prosedürü, safrayı safra kesesinden dışarıdan ileostomi torbasına yönlendiren cerrahi bir yaklaşımdır.
Büyümeyi artırmak için hastalara yağda çözünen vitaminler ve bazen de orta zincirli trigliseritler desteklenmelidir . Karaciğer sentetik disfonksiyonu önemli olduğunda hastalar transplantasyon için listelenmelidir . Bulaşma riskini belirlemek için aile üyeleri PFIC mutasyonları açısından test edilmelidir.
Prognoz
Hastalık tipik olarak ilerleyicidir ve karaciğer nakli yapılmazsa fulminan karaciğer yetmezliğine ve çocuklukta ölüme yol açar . PFIC-2’de hepatoselüler karsinom çok erken yaşta gelişebilir; küçük çocuklar bile etkilendi.
Akraba evliliğinin önemli bir risk faktörü olduğuna inanılmaktadır.
Benzer taşıma proteini mutasyonlarının gebeliğin intrahepatik kolestazı için daha yüksek bir risk oluşturduğuna inanılmaktadır .
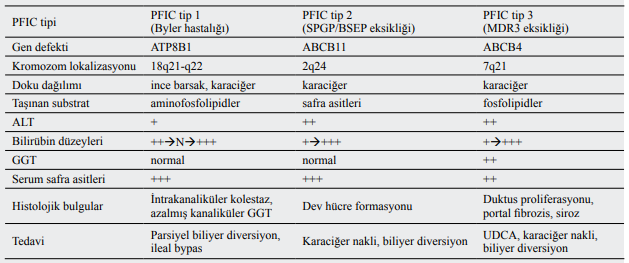
Progresif familyal intrahepatik kolestaz (PFIC), sıklıkla erken süt çocukluğu ve çocukluk döneminde görülen, yaşamın ilk dekadında siroza ilerleyen, otozomal resesif geçişli, nadir bir kolestaz formudur. Hepatobiliyer taşıyıcı proteinleri kodlayan genlerdeki mutasyonlar sonucu gelişmekte ve gerçek sıklığı bilinmemektedir
Başlıca klinik bulguları sarılık, kaşıntı, gelişme geriliği, hepatosplenomegali ve kolestazdır. Ursodeoksikolik asit ve parsiyel eksternal biliyer diversiyon, karaciğer nakline alternatif tedaviler olarak uygulanmaktadır
Son moleküler ve genetik çalışmalarla PFIC’ın üç tipi belirlenmiştir
Kolestaz, karaciğer hastalıklarının sık görülen belirleyici ve ciddi bir bulgusudur. Safra oluşumunu ya da akımını azaltan patolojik duruma kolestaz adı verilir ve safra ile atılması gereken maddelerin vücutta birikmesine neden olan her koşulu içerir. Kolestaz, obstruktif ve hepatoselüler tip olarak ikiye ayrılır. Obstruktif tip kolestazda biliyer sistemde anatomik veya işlevsel tıkanmalar görülür. Biliyer atrezi, koledok kisti, safra taşı, safra kanal darlığı ve biliyer sisteme bası yapan kitle gibi nedenler ekstrahepatik obstruktif tip kolestaza yol açarken; konjenital hepatik fibrozis, Caroli hastalığı, Alagille sendromu, sklerozan kolanjit gibi hastalıklar da intrahepatik obstruktif kolestaza neden olurlar
Kolestaz, safra oluşum ya da atılım mekanizmalarının bozulmasıyla ortaya çıkar. Safra transport bozuklukları, safra asit biyosentez bozuklukları, kolesterol biyosentez bozuklukları, mitokondriyal hastalıklar, ilaç ve toksik etkenler sonucunda gelişebilir
Serum GGT düzeyi, PFIC tip 1 ve 2’de normal, tip 3’te artmış olarak bulunur.
Progresif familyal intrahepatik kolestazın tipik bulguları, yaşamın ilk birkaç ayında ortaya çıkan sarılık, ciddi kaşıntı, hepatomegali, büyüme ve gelişme geriliğidir. Ayrıca ishal ve pankreatik yetmezlik gelişebilir. Süt çocukluğu döneminden erişkin periyoda kadar herhangi bir dönemde siroz gelişebilir (
Progresif familyal intrahepatik kolestaz, değişik etiyolojilerin neden olduğu heterojen bir grup bozukluktur
İlk kez 1965 yılında Clayton ve arkadaşları tarafından Amish soyunda “Byler hastalığı” olarak tanımlanmıştır
Hastalığın patogenezinden safra kanalcıkları düzeyinde safra asidi transportunun bozukluğu sorumlu tutulmaktadır. Safra içeriğinin kanaliküler taşınması genellikle safra akımının hız kısıtlayıcı basamağıdır. Bu taşıyıcı proteinlerin çoğu ATP bağımlı olarak çalışmaktadır. Taşıyıcı proteinlerin genetik ve spesifik ayırımı ile hepatositlerden safra kanaliküllerine farklı taşıyıcı proteinlerin kullanıldığı gösterilmiştir.
Bu taşıyıcı proteinler, safra asitlerini taşıyan safra tuzu salınım pompası (BSEP) ve sister of P glikoprotein (SPGP); organik anyonları taşıyan çoklu ilaç direnci ile ilgili protein 2 (MRP2) ve kanaliküler multispesifik organik anyon taşıyıcısı (CMOAT); fosfolipidleri taşıyan çoklu ilaç direnci ile protein 3 (MDR3) olarak tanımlanmıştır. Bu taşıyıcı proteinleri kodlayan genlerdeki mutasyonlar, fonksiyonlarının bozulmasına, neticede safra asit sekresyonunda veya metabolizmasında defekte yol açmaktadır. Son moleküler ve genetik çalışmalar ile PFIC için sorunlu genler tanımlanmıştır
Bu taşıyıcı proteinleri kodlayan genlerdeki mutasyonlarla PFIC alt tipleri belirlenmiştir
Ayrıca PFIC benzeri karaciğer bulgularına neden olan primer safra asit sentez bozuklukları PFIC tip 4 olarak değerlendirilmektedir
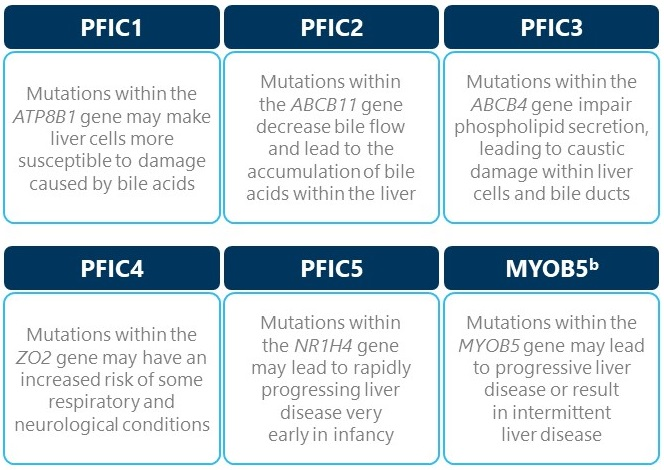
Serum GGT düzeyi, PFIC tip 1 ve 2’de karakteristik olarak normal veya hafif yüksektir ve ciddi kaşıntı görülür. Tip 3’te serum GGT düzeyi yüksek olup belirgin kaşıntı yoktur
PFIC tip 3’te safra yolları korunmuş olmasına rağmen duktular proliferasyon, portal fibrozis ve erken dönemde inflamatuar cevap vardır. MDR3 p-glikoproteindeki mutasyon sonucunda safranın önemli elemanlarından olan fosfolipidlerin atılımında defekt oluşur. Tip 3, hafif kaşıntı, serum safra asitlerinde orta derecede yükseklik ve safrada safra asitlerinin normal olması ile karakterizedir
Labaratuvar bulgularında kolestazı olan olgunun GGT ve serum safra asit düzeyi yüksek olup, karaciğer biyopsisinde safra duktus proliferasyonu, hidropik dejenerasyon ve portal fibrozis izlendi. Progresif familyal intrahepatik kolestazın tedavisinde UDCA ve parsiyel biliyer diversiyon karaciğer nakline alternatif olarak uygulanmaktadır
Ekinci ve arkadaşları inatçı kaşıntısı olan, medikal tedaviye yanıt alamadıkları PFIC’lı iki çocukta parsiyel eksternal diversiyon ile iyi sonuç aldıklarını bildirmişlerdir
UDCA’in tüm PFIC tiplerinde etkili olduğu ileri sürülmüştür. GGT düzeyi yüksek ve normal iki PFIC grubu oluşturularak yapılan bir çalışmada, 20-30mg/kg/g do zunda 2-4 yıl uygulanan UDCA tedavisiyle GGT yüksek grupta daha fazla olmak üzere her iki grupta da iyi yanıt alındığı bildirilmiştir
Ursodeoksikolik asit, kolestatik karaciğer hastalığında endojen safra asitlerinin hepatositlerden atılımını arttırır ve barsaktan emilimini engelleyerek tekrar karaciğere dönüşünü sınırlar. Endojen safra asitlerinin yoğunlaşmasının azalmasıyla hepatosit fonksiyonları düzelir. Ayrıca kanaliküler/bazolateral transporterların salınımını arttırır. Gama-glutamil transferaz düzeyi yüksek PFIC’lı vakalarda UDCA kullanımı safra asitleri bileşenlerinin hidrofilik safra asitleri lehine düzenlenmesi konusunda da ek yarar sağlar
Progresif familyal intrahepatik kolestazlı hastaların başlangıç tedavisinde UDCA ve parsiyel eksternal biliyer diversiyon dikkate alınmalı ve bu tedavilerde yetersizlik düşünüldüğünde karaciğer nakli planlanmalıdır.