



Aşağıda sık görülen immün yetmezlik ve o hastalığa ait karakteristik klinik özellik eşleştirmelerinden hangisi yanlıştır?
A) Bruton agamaglobulinemisi: Tonsil dokusunun olmaması veya çok küçük olması
B) Selektif Ig A eksikliği: Sprue benzeri tablo
C) Wiskott-Aldrich sendromu: İri, sayıca az trombosit ********************
D) Hiper IgE sendromu: Kaba yüz görünümü
E) Kronik granülomatöz hastalık: Tekrarlayan abseler
Wiskott-Aldrich sendromu 1 yaş civarında küçük ve sayıca az trombositler, sekonder purpura ve atopik dermatit triadı ile karakterizedir
Wiskott–Aldrich sendromu (WAS), egzama, trombositopeni (düşük trombosit sayısı), immün yetmezlik ve kanlı ishal (trombositopeniye sekonder) ile karakterize, X’e bağlı, nadir görülen bir resesif hastalıktır.
Aldrich’in 1954’teki orijinal tanımına uygun olarak bazen egzama-trombositopeni-immün yetmezlik sendromu olarak da adlandırılır.
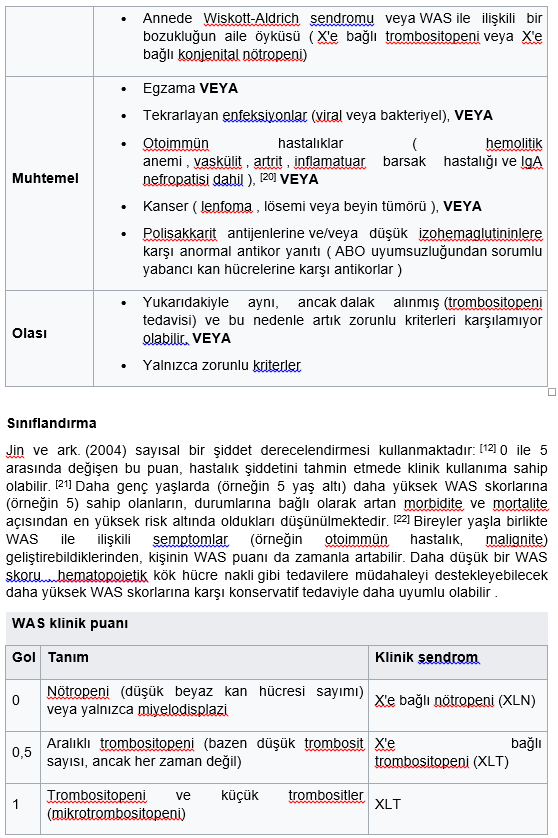
WAS ile ilişkili X’e bağlı trombositopeni (XLT) ve X’e bağlı konjenital nötropeni (XLN) bozuklukları benzer ancak daha az şiddetli semptomlarla ortaya çıkabilir ve aynı genin mutasyonlarından kaynaklanır.
Wiskott-Aldrich sendromu olarak adlandırılan gendeki (WAS geninin kısaltması, gen lokusu Xp11.23-p11.22) mutasyonlar, hücresel sinyal iletiminin bozulmasına yol açar. Aktin polimerizasyonu sınırlıdır, bu da megakaryositlerden trombosit oluşumunu azaltır .
Doğumdan kısa bir süre sonra yenidoğanda trombositopeni sonucu ilk noktasal (peteşial) kanama meydana gelir , daha sonraki yıllarda ise gastrointestinal ve intrakranyal kanamalar meydana gelir. Tipik egzama da atopik dermatite benzer şekilde yaşamın ilk günlerinde gelişir.
T hücresi bağışıklığı başlangıçta hala normal iken, yaşamın ilk yılında humoral bağışıklık önemli ölçüde azalır. T hücresi bağışıklığı önümüzdeki birkaç yıl içinde sürekli olarak azalır ve bu da enfeksiyona duyarlılığı artırır. Pnömoni , orta kulak iltihabı , septisemi ve menenjit gibi tekrarlayan fırsatçı enfeksiyonlar ilk kez yaşamın ikinci yılında ortaya çıkar . Yaygın patojenler arasında pnömokoklar , Haemophilus influenzae , meningokoklar ve Pneumocystis jirovecii yer alır .
Genç hastalarda sıklıkla vaskülit , artrit ve hemolitik anemi gibi otoimmün hastalıklar aynı anda görülür . Wiskott-Aldrich hastalarında lenforetiküler sistem maligniteleri daha sık görülür.

Aşağıdaki karakteristik değişiklikler Wiskott-Aldrich sendromu şüphesini artırmaktadır:
1-IgM antikorları düşük, IgG antikorları normal, IgA, IgD ve IgE normun üzerinde yükselmiştir .
2-Kan sayımı tipik olarak küçük kan trombositleriyle birlikte ciddi trombositopeni gösterir (düşük ortalama trombosit hacmi = MPV).
3-Aşı antikorları azalır.
4-Şiddetli lenfopeni ilk kez altı yaşından sonra ortaya çıkar .

Wiskott-Aldrich sendromunda Wiskott-Aldrich proteini (WASP) düzgün şekilde üretilmez. WASP hücre iskeletinin organizasyonu için gereklidir . Hematopoietik kök hücrelerde protein eksikse , T ve B lenfositleri, makrofajlar , dendritik hücreler , NK hücreleri ve trombositler tam olarak işlevsel değildir. Tanıyı doğrulamak için belirlenebilir.
Tanı, mutasyon analizi kullanılarak yapılan moleküler genetik inceleme ile doğrulanır. Doğum öncesi tanı da mümkündür.
Wiskott-Aldrich sendromunda karakteristik bulgular; Trombositopeni, Egzama ve Rekürren enfeksiyonlardır.
—————————-
Wiskott–Aldrich sendromu için aşağıdaki ifadelerden hangisi yanlıştır?
A) X’ bağlı resesif kalıtılır
B) Trombositopeni görülebilr ve bu trombositler makrotrombosit ile karakterizedir***
C) Genellikle altıncı aydan sonra ortaya çıkan yineleyen pyojenik infeksiyonlar mevcuttur
D) Çocuklarda egzema benzeri cilt lezonları görülebilir
E) Azalmış Ig M, artmış Ig A ve Ig E düzeyleri görülebilir
Cevap B
Wiskott–Aldrich sendromunda trombositopeni görülebilir ve bu trombositler mikrotrombositopeni ile karakterizedir.
Wiskott–Aldrich sendromu: X’e bağlı resesif geçiş gösterir. Tüm hematopoetik kökenli hücrelerde bulunan Wiskott–Aldrich sendrom proteini (WASP) kodlayan gende mutasyon mevcuttur. WASPà hücre iskeletinin yapılanmasında ve sinyal iletiminde önemli role sahiptir. Konjenital trombositopeni, mikrotrombositlere bağlı kanama
Genellikle altıncı aydan sonra ortaya çıkan yineleyen pyojenik infeksiyonlar mevcuttur. Bir yaş civarında ekzema görülür. Serum IgM düşük, Ig A ve Ig E düzeyleri artmış saptanabilir.
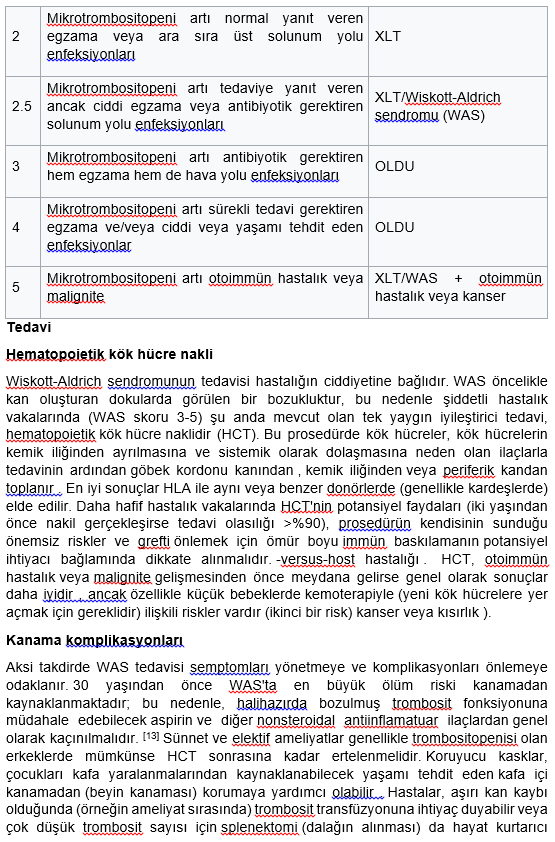
Tedavisi KİT


Trombositopeni, ekzema ve immün yetmezlik ile giden, T lenfositlerin progresif azalması ile karakterize immün yetmezlik hastalığında en olası gen mutasyonu hangisidir?
A) BAFF
B) 22q11 delesyonu
C) WASP************
D) CD40 ligand
E) IL-7 reseptör geni
Wiskott-Aldrich Sendromu:
X’e bağlı geçiş gösteren, WAS proteinini (WASP) kodlayan gende mutasyon sonucu oluşur.
Karakteristik olarak trombositopeni, egzema ve immün yetmezlik tablosu gelişir.
Timus başlangıçta normal görünümdedir fakat hastalık ilerledikçe T hücre sayısı progresif azalır ve lenf nodlarında parakortekste incelme görülür.
Polisakkarit antijenlere antikor yapımı yoktur. IgM düşük, IgA ve IgE yüksek, IgG normal seviyelerdedir.
B hücreli lenfoma riski artmıştır.

Aşağıdaki primer immun yetersizlik sendromlarından hangisi immun yetmezliğe eşlik eden kanama diatezi
ve konjenital trombositopeni ile karakterizedir ?
A) Hiper IgE sendromu
B) Kronik granülomatöz hastalık
C) X’e bağlı agammaglobulinemi
D) Wiskott-Aldrich sendromu
E) Ataksi-Telejiektazi sendromu
Doğru Cevap: D
Primer immun yetersizlikler içerisinde konjenital trombositopeni nedeniyle kanama diyatezinin görüldüğü immun yetmezlik Wiskott-Aldrich sendromudur. Wiskott_Aldrich sendromunda trombosit sayısının düşüklüğü yanında mikrotrombositler, trombosit agregasyon defektleri de vardır. Serum IgM düzeyi genellikle düşüktür. Kombine bir immune yetersizliktir. Tedavisi kemik iliği naklidir.
Canlı aşılar kullanılmamalıdır.