İNCE MEMBRAN HASTALIĞI (İYİ HUYLU AİLEVİ HEMATÜRİ),
Tekrarlayıcı veya devamlı olan hematüri ile seyreden bir hastalıktır. Genellikle proteinüri (idrarda protein) yoktur varsa da çok düşük miktardadır. Böbrek dışı bulgular görülmez. Tanı için böbrekten parça alınması gerekebilir.
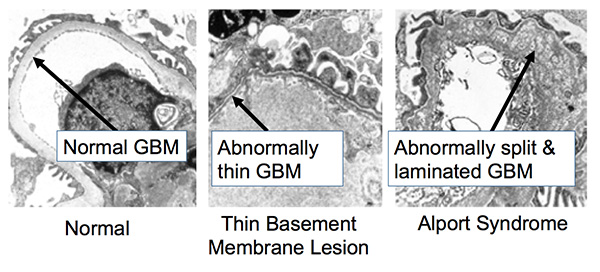
Glomerül bazal membranın normalde olması gereken 300-400 mikron kalınlıktan 150-225 mikrona inmesi ile karakterize ailevi asemptomatik hematüri olarak bilinen, selim gidişli bir hastalıktır.
Hastalarda, hafif ya da orta derecede proteinüri bulunsa da böbrek fonksiyonları normaldir ve prognoz son derecede iyidir. Bazı hastalarda tip IV kollajenin a3 ve a4 zincirlerini kodlayan genlerde mutasyon izlenmiştir

İnce Membran Hastalığı ince bazal membran hastalığı; ince bazal membran nefropatisi; ince membran nefropatisi; iyi huylu ailesel hematüri;
İnce bazal membran hastalığının moleküler temeli henüz tam olarak aydınlatılamamıştır; ancak bazı ailelerde tip IV kollajende kusurlar rapor edilmiştir. İnce bazal membran hastalığı olan çoğu hastanın yalnızca güvenceye ihtiyacı vardır. Aslında bu hastalık, genellikle iyi huylu seyretmesi nedeniyle daha önceleri “iyi huylu ailesel hematüri” olarak adlandırılıyordu. Kontrollü çalışmalar eksik olmasına rağmen, anjiyotensin dönüştürücü enzim inhibitörlerinin hematüri ataklarını azalttığı öne sürülmüştür.
Birlikte var olan hiperkalsiüri ve hiperürikozürinin tedavisi de hematürinin azaltılmasında yardımcı olacaktır. İnce bazal membran hastalığının moleküler temeli henüz tam olarak aydınlatılamamıştır; ancak bazı ailelerde tip IV kollajenin a4 zincirini kodlayan gendeki kusurlar rapor edilmiştir. COL4A3’ün yeni bir mutasyonu, Alport sendromuna ve ince bazal membran nefropatisine farklı bir katkı sunar”.

“Daha önce Alport sendromunda açıklanan ince bazal membran hastalığında COL4A4 mutasyonu”. Kollajen IV ile İlgili Nefropatiler (Alport Sendromu ve İnce Bazal Membran Nefropatisi) İnce bazal membran hastalığına genellikle otozomal ilerleyici olmayan dominant paternde COL4A3 ve COL4A4 genindeki Heterozigot mutasyon neden olur ve X kromozomunda COL4A5 genindeki heterozigot mutasyon kadınlarda İnce bazal membran hastalığına neden olabilir.
Diğer ismi ile benign familyal familyal hematüri. glomerüler bazal membranı diffüz olarak incelmiştir. persistan mikroskopik hematüri yapar. epizodik gros hematüriyi özellikle üsye sonrası yapabilir. semptomları ıga nefropatisi’ne benzer. hafif proteinüri olabilir. renal fonksiyonlar normaldir. kby’e dönüşüm çok nadirdir.
ince glomerüler bazal membran hastalığında tekrarlayan hematüriler olur. zaten diğer adı da benign familyal hematüri’dir. od kalıtılır bu hastalık. ve hem mikroskopik hem de makroskopik hematüri yapabilir. ve bunlarda prograsif bir böbrek hasarı yok yani kalıcı bir böbek hastalığı yok.
Burada aslında tip4 kollajen defekti olur. bunlarda proteinüri beklenmez.
Tanı aile öyküsü ile konur gerekmedikçe böbrek biyopsisi yada genetik inceleme yapılmaz.
Alport Syndrom und Nephropathien vom Typ der dünnen Basalmembran: COL4A3, COL4A4, COL4A5, FN1, CD151, MYH9 (6 Gene).“
Ein anderes Labor untersucht im Subpanel Alport Syndrom und Nephropathien vom Typ der dünnen Basalmembran die sieben Gene COL4A3, COL4A4, COL4A5, FN1, CD151, MYH9 und PXDN.
: Nephropathie vom Typ der dünnen Basalmembran