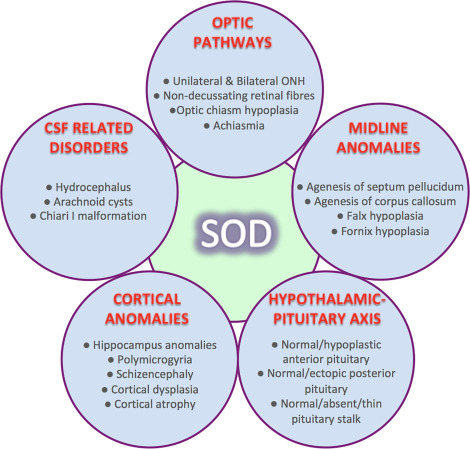
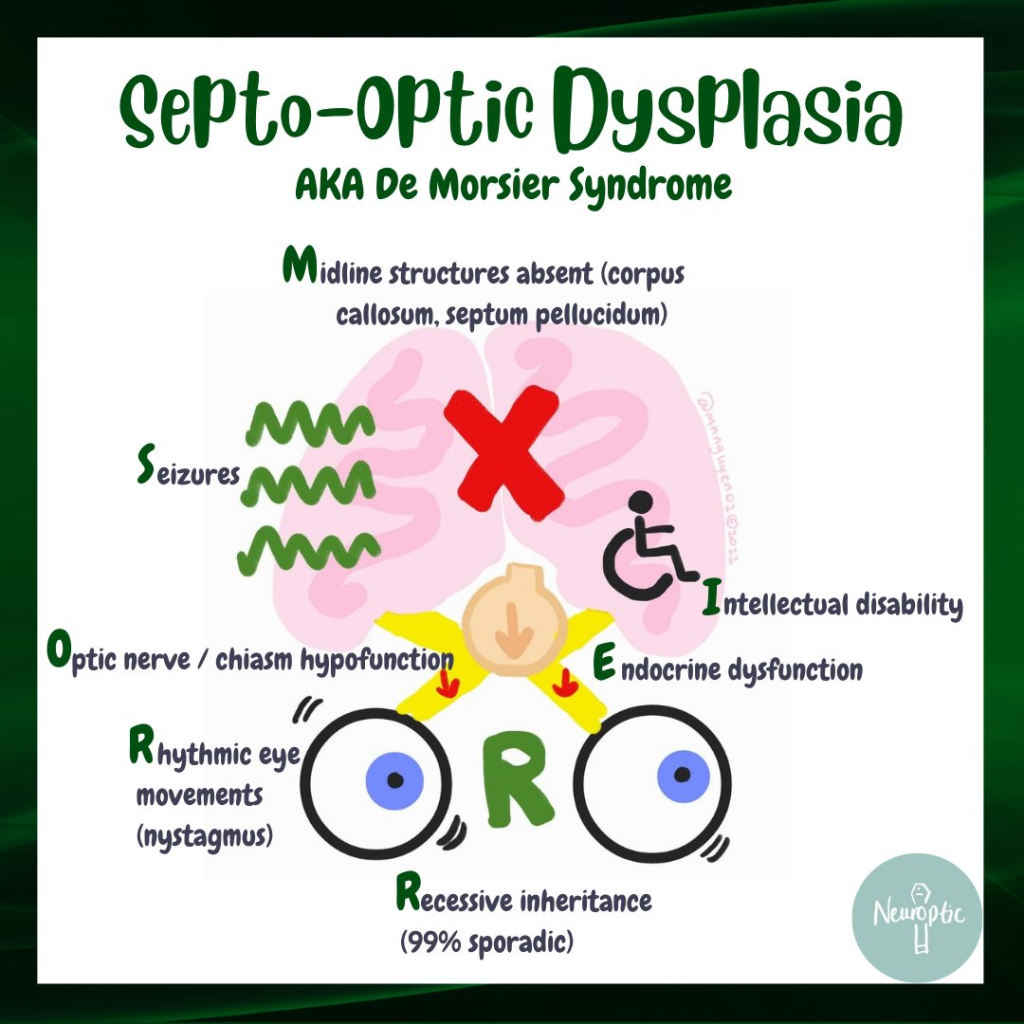
Septa Optik Displazi, De Morsier Sendromu olarak da bilinen nadir bir konjenital durumdur. Sendromun klasik triadındaki bulgular; beyin orta hat yapılarında anomaliler, optik sinir hipoplazisi ve hipofiz hormon disfonksiyonlarıdır. Triaddaki bulguların en az ikisinin bulunması ile tanı konulur.
Optik sinir hipoplazisi, hipofizer yetmezlik veya beyin orta hat yapılarda anomali saptanması durumunda triada ait diğer bulguları araştırmalıdır ve gereğinde hormon replasman tedavileri başlanmalıdır.
Septaoptik Displazi (SOD); Septum pellicidum agenezi ve optik sinir anomalisi olan bir vaka aracılığı ile ilk olarak 1941 yılında Reeves tarafından tanımlandı ve 1956 yılında de Morsier tarafından Septa Optik Displazi olarak adlandırıldı
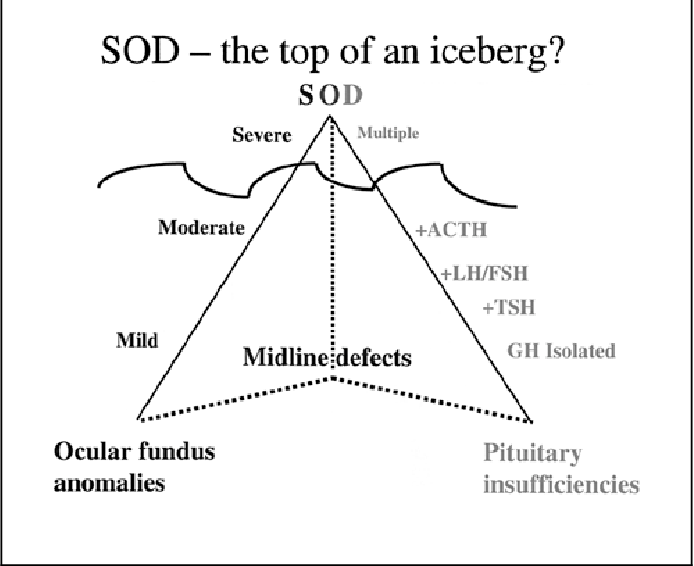
Daha sonra tanımlanan hastalarda ilave olarak hipotalamo–pituiter aksın disfonksiyonundan kaynaklanan endokrin anormalliklerin de eşlik ettiği gösterildi. SOD; nadir bir konjenital durumdur. Klasik triadındaki bulunan optik sinir hipoplazisi, hipotalamik/pituiter hormon anormallikleri ve beyin orta hat yapılarda septum pellicidum ve/veya korpus kallosum agenezisi bulgularından 3’ünden ikisinin bulunması ile tanı konulabilir. SOD fenotipi oldukça heterojendir. Sadece %30 kadar vakada triada ait tüm bulgular mevcuttur. SOD vakalarının çoğunluğu sporadiktir.
Optik sinir hipoplazisi (ONH) sendromun en sık ve genellikle ilk bulgusudur. Sıklıkla bilateraldir. Olguların çoğunda görme keskinliğinde azalma, nistagmus ve astigmatizm mevcuttur. Beyin orta hat anomalileri septum pellicidum yokluğu ve korpus kallosum agenezi başta olmak üzere forniks aplazisi, şizensefali, serebellar hipoplazi, araknoid kistler gibi geniş bir yelpazeyi içerir
Pituiter hipoplazi kendini izole hormon yetersizliği şeklinde gösterebildiği gibi panhipopituitarizm tablosu ile de karşılaşılabilir. En çok GH yetersizliği olmakla birlikte daha az oranda ACTH ve TSH yetersizlikleri saptanır. Gonadotropik fonksiyonlar genellikle korunmuştur.

Septaoptik Displazi (SOD); hipopituitarizmi olan çocukların %50’sinden çoğunda saptanan, heterojen bir grup bozukluğa işaret eden bir genetik sendromdur. Bu sendromda septum pellicidum agenezisi, çeşitli derecelerde görme bozukluğuna yol açan optik sinirde hipoplazi veya aplazi, tersiyer hipopituatirizme sebep olan hipotalamus abnormalitesi mevcuttur. %30 vakada hemen tüm klinik bulgular gözlenir iken, %60 hipopituitarizm ve %60 septum pellicidum agenezi gözlenir. Klinik bulgular; çeşitli derecelerde pituiter yetmezlik, değişik düzeylerde psikomotor retardasyon, hafiften ciddiye değişen görme bozukluğu, termoregulatuar bozukluklar, konjuge hiperbiluribinemi ve nöbetler şeklinde ortaya çıkabilir. Hipotalamik pituiter aksın fonksiyon bozukluğundan kaynaklanan en sık endokrin anormallik büyüme gelişme yetersizliği olup, GH/IGF1 aks anormalliği bu duruma neden olmaktadır
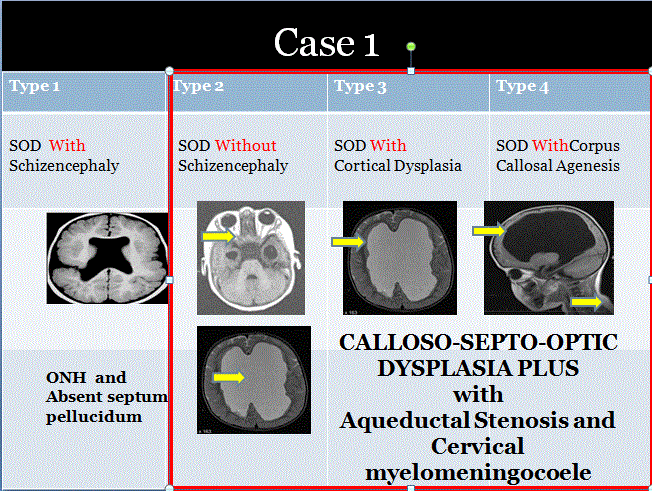
Tanı genellikle erken yaşta konulur. Büyüme gelişme geriliği nedeniyle başvuran hastalarda nadir de olsa SOD olabileceği düşünülmelidir. Ülkemizde Akyürek ve ark büyüme gelişme geriliği ile başvuran 2 olgu bildirmiştir. Carman ve ark. tarafından da pakigiri, şizensefali ve diabetes insipitus tespit edilen septooptik displazili bir vaka bildirilmiştir.
Hipofizer hormon yetersizliği olarak en sık GH yetersizliği görülen sendromda ACTH ve TSH eksiklikleri ikinci ve üçüncü sıralardadır. Sendromda gonodotropin fonksiyonlarının genel olarak korunduğu bildirilmiştir.

De Morsier Sendromu büyüme geriliği ve optik disk kolobomu ile gelen vakalarda endokrin tablonun ortaya konması gerekliliği önemlidir; SOD tanısı için mutlaka beyin ve hipofiz MR görüntülemeleri yapılması, ancak görüntüleme ile patoloji saptanmaması durumunda SOD tanısının dışlanmayacağının bilinmesi; hipogonadotropik hipogonadizm durumlarında ayırıcı tanı sonrası erken hormon replasman tedavisi başlanarak seksüel gelişim ve osteoporoz açısından olumsuz gelişmelerin önlenmesini sağlamakdır. Göz muayenesi, hipofiz hormon profili ve beyin görüntüleme yöntemleri tanı için yeterli olup replasman ve destek tedavi ile bireylerin fiziksel ve mental durumlarında en yüksek performansa ulaşılmaya çalışılmalıdır.
De Morsier sendromu olarak da bilinen septo-optik displazi ( SOD ), optik sinirin az gelişmişliği , hipofiz bezi fonksiyon bozukluğu ve septum pellusidumun (beynin orta hat kısmı) yokluğunun bir kombinasyonunu içeren nadir bir konjenital malformasyon sendromudur . ). Klinik tanı için bu özelliklerden iki veya daha fazlasının mevcut olması gerekir; hastaların yalnızca %30’unda bu üç özellik birden bulunur.
Fransız-İsviçreli doktor Georges de Morsier, ilk kez 1956’da, gelişmemiş veya eksik septum pellucidum ile optik sinirlerin hipoplazisi ve kiazma arasındaki ilişkiyi fark etti .
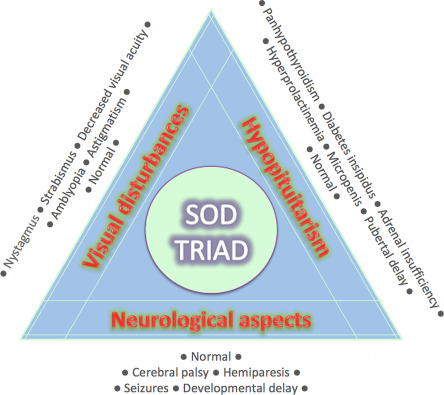
SOD semptomları, optik sinir az gelişmişliği, hipofiz hormonu anormallikleri ve orta hat beyin anormallikleri ile ilgili olanlara ayrılabilir. Semptomların şiddeti büyük ölçüde değişebilir.
Optik
SOD’lu kişilerin yaklaşık dörtte birinde, optik sinirlerin az gelişmesinin bir sonucu olarak bir veya her iki gözünde önemli görme bozukluğu vardır. Gelişimsel gecikmeler, iki taraflı optik sinir hipoplazisi olan çocuklarda, tek taraflı optik sinir hipoplazisi olanlara göre daha yaygındır. [6] Bilateral optik sinir hipoplazisi de daha ciddi bir hastalık seyri ile ilişkilidir.
Nistagmus (istemsiz göz hareketleri, genellikle yan yana) olabilir . Bilateral optik sinir hipoplazisi vakalarında bu genellikle yaşamın ilk üç ayında tespit edilebilir. Bunu ilk yıl gelişen şaşılık takip edebilir .
Hipofiz
SOD’da hipofiz bezinin az gelişmiş olması , çoğunlukla büyüme hormonu eksikliği şeklinde hipopitüitarizme yol açar . Ağır vakalarda panhipopituitarizm ortaya çıkabilir.
Orta hat
SOD’da korpus kallozum ve septum pellucidum gibi orta hat beyin yapıları normal şekilde gelişemeyebilir ve bu da nöbetler veya gelişimsel gecikme gibi nörolojik sorunlara yol açabilir. [8] Nöbet geçiren hastaların kortikal displazi , polimikrogiri ve şizensefali gibi ek nörolojik anormallikler gösterme olasılığı daha yüksektir . Bu tür anormallikler her zaman spastik kuadripleji mevcut olduğunda tanımlanır.
Nörolojik semptomlar tipik olarak SOD’un geç başlangıçlı belirtileri olarak kabul edilir. Yaygın başlangıç belirtileri arasında epilepsi , gelişim gecikmeleri ve uzuv zayıflığı yer alır. Entelektüel yetenekler normalden ciddi zihinsel engelliliğe kadar geniş bir yelpazede değişiklik gösterir . İlk çalışmalar , vakaların %71’inde zihinsel engellilik , %57’sinde serebral palsi ve %20’sinde davranış sorunları oluştuğunu gösterdi; ancak daha ileri araştırmalar, bu semptomların daha az yaygın olabileceğini ve ek nörolojik anormalliklerden kaynaklanabileceğini gösterdi.
Nedenler
SOD, hamileliğin 4-6. haftalarında embriyonik ön beyin gelişimindeki anormallikten kaynaklanır. SOD’un bilinen tek bir nedeni yoktur ancak hem genetik hem de çevresel faktörlerin rol oynayabileceği düşünülmektedir.
Genetik
En az bir genetik formun ( HESX1 ) varlığını düşündüren nadir ailesel nüks rapor edilmiştir .
Beş homozigot ve sekiz heterozigot patojenik HESX1 mutasyonu keşfedilmiştir. Homozigot mutasyonlara sahip hastalar tipik bir SOD fenotipine sahipken, heterozigot mutasyonlara sahip hastalar hafif derecede etkilenir.
SOD’da HESX1’in yanı sıra OTX2, SOX2 ve PAX6’daki mutasyonlar da suçlanmıştır.

SOD hastalarındaki SOX2 mutasyonları, mikroftalmi ve anoftalmi gibi ciddi iki taraflı oküler anomalilerle ilişkilidir . SOX2 mutasyonlarıyla ilişkili ek özellikler arasında gelişimsel gecikme , özofagus atrezisi , kısa boy ve sensörinöral işitme kaybı yer alır . Genetik anormallikler hastaların yüzde birinden daha azında tanımlanır.
Teşhis
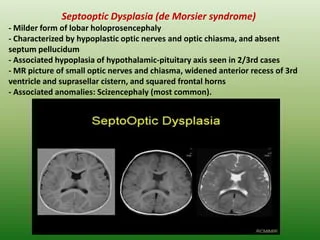
Aşağıdaki üçlüden en az ikisinin mevcut olması durumunda SOD tanısı konur: optik sinirin az gelişmişliği; hipofiz hormonu anormallikleri; ve orta hat beyin anormallikleri. Tanı genellikle doğumda veya çocukluk döneminde konur ve klinik tanı MRI taramaları ile doğrulanabilir .
Tedavi
SOD’un tedavisi yoktur. Tedavi semptomatiktir ve nörologlar , oftalmologlar ve endokrinologları içeren multidisipliner bir uzman ekibi gerektirebilir . Hormon eksiklikleri HRT ile tedavi edilebilir ancak görme bozuklukları genellikle tedavi edilemez.