


Konjenital aplastik anemi…Fanconi anemisi
Hiperpigmentasyon, başparmak yokluğu ve mikrosefali bulunan 8 yaşındaki kısa boylu çocukta pansitopeni var, tanı…Fanconi anemisi
Tanı için yapılacak test…Diepoksibutan (DEB) testi veya Mitomisin C testi
Fanconi anemisinde ilk ortaya çıkan hematolojik bulgu…Trombositopeni
Fanconi anemisinde kemik iliği…Hiposellüler ve yağlı

Artan malign hastalık riski…AML,MDS,Cilt-Dil-Baş-boyun Mide-Özefagus kanserleri
Fanconi anemisi tedavisinde kullanılanlar…Androgen,Steroid,GM-CSF, KİT
Fanconi anemisinde ortalama kemik iliği bulguları yaşı…8 yaş

Fanconi Aplastik Anemisi kromozomal kırılma sendromlarından biridir ve bu hastalıkta lösemi riski artmıştır.
Hâlbuki renal proksimal tübül hasarı ile karakterize olan Fankoni sendromunun lösemi ile ilişkisi yoktur.
Fanconi anemisi (FA) kalıtsal KİY sendromları arasında en sık görülenidir. Fanconi anemisi genetik ve fenotipik olarak oldukça heterojen olmakla birlikte zaman içinde gelişen kemik iliği yetmezliği, artmış maliyn hastalık riski ve konjenital malformasyonlar en önemli özellikleridir
FA, Myelodysplastic sendrom ve AML ile beraber olabilir.
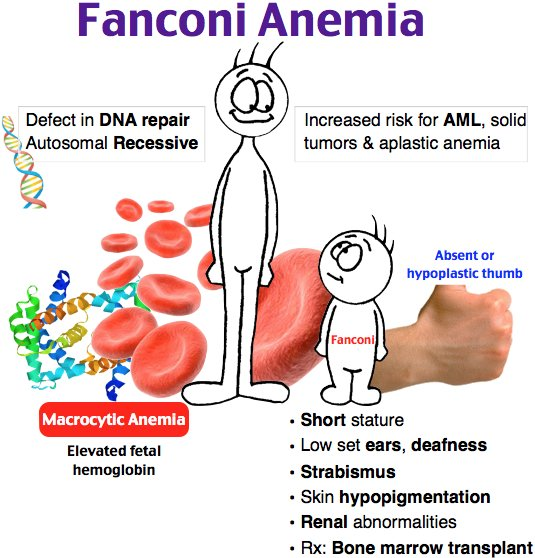
Kalıtım özelliği sıklıkla otozomal resesif karakterdedir. Ancak bu zaman kadar tanımlanmış olan 13 farklı genden sadece birinde kalıtsal geçiş X’e bağlı resesif özelliktedir. Aile içi evliliklerin daha çok olduğu ülkemizde bu nedenlerle batı toplumlarından daha sık görülmektedir. Fanconi anemisinde hematolojik bozukluklar genellikle 5 ila 10 yaş (aralık: 1 ila 31 yaş) arasında ortaya çıkar. Uluslararası “Fanconi Anemisi Kayıt Sistemi (IFAR)” verilerine gör FA hastalarında 40 yaşına kadar kemik iliği yetmezliği, hematolojik malinite ve diğer malinitelerin ortaya çıkma riski sırası ile %90, %33 ve %28’dir.
Konjenital malformasyonlar arasından en önemlileri olarak değişik tarzlarda başparmak anomalisi, mikrosefali, mikroftalmi, ciltte pigmentasyon değişiklikleri, kalp ve böbrek anomalileri sayılabilir
Ancak FA olan hastaların yaklaşık %25 ila %40’ında hiçbir anomali tespit edilemez . Bu nedenlerle özellikle çocuklarda veya genç erişkinlerde kemik iliği yetmezliği bulguları olduğu her durumda fiziksel anomali olup olmadığına bakılmaksızın FA ön tanısı ile gerekli laboratuar incelemeleri yapılmalıdır. Fanconi anemisinin tanısı için ilk olarak diepoksibütan (DEB) veya mitomisin-C (MMC) ile uyarılmış periferik kan lenfositlerinde kromozom kırıklarının artmış olduğunun gösterilmesi gereklidir.
Bazı ender FA olgularında lenfosit popülasyonundaki somatik mozaikizim nedeni ile bu test sınırda pozitif veya negatif olabilmektedir. Bu nedenle kan lenfositleri ile kromozon kırılma incelemesi normal değerlendirilmesine karşın klinik açıdan kuvvetle FA düşünülen durumlarda DEB veya MMC ile kromozom kırılma incelemesinin cilt fibroblastlarıyla yapılması gerekmektedir.
Koromozom kırılma incelemesinin kesin tanımlayıcı olamadığı durumlarda FANCD2 monoubikitasyonunun saptanması alternatif olarak önerilebilir.
Fanconi anemisi tanısı konduktan sonra hastanın kan değerlerinin yakın takibinin yanı sıra yılda en az bir kez kemik iliği incelemesi yapılarak morfolojik açıdan dizplazi gelişimi ve özellikle de klonal sitogenetik bozuklukların (7-, 7q-, trisomi 8 gibi) gelişimi açısından takip edilmelidir. Fanconi anemisine bağlı gelişen sitopenilerde androjen tedavisi ile kan değerlerinde yükselme sağlanabilse de bunun tüm hastalar için geçerli olmamasının yanı sıra bu olumlu etkinin zaman içinde kaybolması nedeni ile kalıcı bir tedavi yöntemi olarak kabul edilmemektedir.
FA hastalarında kemik iliği yetmezliği için günümüzdeki tek küratif tedavi seçeneği HKHT’dir. FA hastalarında gelişebilen MDS ve lösemilerin prognozu her durumda çok kötü olmakla birlikte en iyi tedavi seçeneğini HKHT oluşturmaktadır.
Fankoni Anemisi: Otozomal resesif geçişli DNA onarım bozukluğunun olduğu bir hastalıktır. Hastalık en sık konjenital aplastik anemi nedenidir. Hastalıkta karakteristik diğer bulgular:
Dalak ve böbrek hipoplazisi + el 1. parmağında hipoplazi ve hiperpigmentasyon
Yetişkin başlangıçlı aplastik anemilerde %5-10 oranında telomeraz defekti saptanmıştır.