Tirozinemi Tip II (Richner Hanhart Sendromu) (Okulokütanöz Tirozinemi)
– Otozomal resesif geçişlidir.
– Tirozin transaminaz (aminotransferaz) eksikliğii vardır.
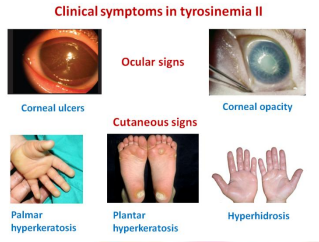
– Göz ve cilt bulgularının olduğu tirozin metabolizma bozukluğudur. Gözlerde aşırı sulanma, kızarıklık, ağrı,fotofobi, herpetiform korneal ülserler ve cilt lezyonları görülür. Ciltte palmar ve plantar punktat hiperkeratoz görülür. Göz lezyonları yaşamın ilk aylarında, cilt lezyonları ise daha sonra ortaya çıkar.
– Mental retardasyon vardır.
– Self mutilasyon görülebilir.
– Hipertirozinemi ve tirozinüri vardır.
–Tirozinemi tip l’den farklı olarak karaciğer, böbrek fonksiyonları ve diğer aminoasidlerin serum konsantrasyonları normaldir.
– Tirozin ve fenilalaninden kısıtlı diyet verilmesiyle klinik ve laboratuar bulgular hızla düzelir. Diyet erken dönemde başlanırsa mental retardasyon gelişimi önlenebilir.
—————————–
Tirozinemi
Tirozinemi çekinik genler ile çocuğa aktarılan, bir protein yapıtaşı olan Tirozin amino asidi metabolizmasındaki çeşitli enzimlerin yeterli çalışmayışı sebebiyle ortaya çıkar.
– Tirozinemi Tip I: Fumarilasetoasetik hidrolaz enzimi yetersizliği sebebiyle ortaya çıkar.
– Tirozinemi Tip II: Para-hidroksifenilpürivik asit dioksijenaz yada diğer adı ile hepatik tirozin aminotransferaz enzimi eksikliği sebebiyle ortaya çıkar.

Vücut, tirozin yapmak için başka bir amino asit olan fenilalanini de kullandığından, fenilalaninin gereğinden fazla alınması da problemlere yol açmaktadır. Bu nedenle, Tirozinemi hastalarının aldıkları protein miktarını ciddi olarak sınırlamaları gerekmektedir. Tirozinemi hastalığının tedavisi yaşam boyu fenilalanin ve tirozin içeriği düşük diyet tedavisidir.
Aşağıdakilerden hangisi Tirozinemi tip 2 için yanlıştır?
A) Fumaril asetoasetat hidrolaz enzimi eksiktir *********
B) Mental retardasyon
C) Palmar plantar keratoz
D) Korneada herpetik lezyonlar
E) İdrarda süksinil aseton artışı yoktur
Tirozin aminotransferaz eksikliğine bağlı nadir otozomal resesif bir hastalıktır.
Mental retardasyon (hafif-orta düzeyde, < %50 görülür), palmar ve plantar punktat hiperkeratoz (ağrılı, kaşıntısız plak) ve herpetiform korneal ülserasyonlar (fluoresin zayıf boyanır, bilateraldir) ile karakterizedir.
Tirozinemi tip I’in aksine süksinilaseton düzeyi, karaciğer ve böbrek fonksiyonları normaldir.
Tedavide phe ne tirozinden kısıtlı diet hastanın kliniğini normalize eder.
TİROZİNEMİ TİP 2 (OKÜLOKUTANÖZ TİROZİNEMİ) diğer adı ile Richner-Hanhart sendromu
Tirozin aminotansferaz (TAT) enzim eksikliğine bağlı gelişir.
Kromozom 16q22.1-q22.3 TAT gen mutasyonu bağlı ortaya çıkar.
Hastalık anneden ve babadan otozomal resesif olarak kalıtılır.
Palmoplantar hiperkeratoz denilen el içi ayak tabanında deri bulguları, gözde herpetiform korneal ülserler denilen karneal etkilenme ve zeka etkilenimi gözlenir.
Tip II tirozinemi, TAT geni tarafından kodlanan tirozin aminotransferaz enziminin eksikliğinden kaynaklanır . Tirozin aminotransferaz, tirozini böbrekler tarafından atılan veya enerji üreten reaksiyonlarda kullanılan daha küçük moleküllere dönüştüren beş enzim serisinin ilkidir .

Bozukluğun bu formu gözleri, cildi ve zihinsel gelişimi etkileyebilir. Semptomlar genellikle erken çocukluk döneminde başlar ve aşırı gözyaşı , ışığa karşı anormal hassasiyet ( fotofobi ), göz ağrısı ve kızarıklık ve ağrılı cilt lezyonlarını içerir.
Avuç içi ve tabanlardaki döküntü tipiktir.
Tip II tirozinemili bireylerin yaklaşık yarısı aynı zamanda zihinsel engellidir.
Tip II tirozinemi, 250.000 kişide 1’den az görülür
Karaciğer ve böbrek tutulumu olmaz.
Göz bulguları ilk aylarda, cilt bulguları 1 yaş civarında görülür.
Belirgin hipertirozinemi görülür (plazma tirozin seviyesi 500 µmol/L üzerindedir).
İdrarda p-hidroksifenilpiruvat, p-hidroksifenillaktat, p-hidroksiasetat artmıştır. N-asetiltirozin, 4- tiramin de idrar organik asitlerinde artmış olabilir.
Tedavide tirozin ve fenilalanin kısıtlı diyet önerilir.

Tedavi edilmemiş annelerin bebeklerinde, mikrosefali ve gelişim geriliği tanımlanmıştır (maternal tirozinemi).