MEN-4 Sendromu: Klinik özellikleri MEN-1’e benzer ancak bu sendromda p27’yi kodlayan CDKN1B mutasyonları vardır. Hastalarda primer hiperparatiroidi (en sık), anterior hipofiz adenomları, gastrik veya bronşiyal karsinoid tümör, Zollinger- Ellison sendromu görülebilir. Ayrıca adrenal, renal ve reprodüktif organ tümörleri görülebilir.
Multiple endokrin neoplazi (MEN), bir hastada en az 2 endokrin organda görülen tümörlerden oluşmaktadır. Spesifik organlarda bulunan tümörlere göre MEN Tip 1-4 olarak sınıflandırılmaktadır. Bu grup hastalıklar otozomal do- minant kalıtılmakla birlikte aile hikayesi olmadan, sporadik olarak da gelişebil- mektedir
En sık görüleni MEN Tip 1 olup, diğer adı Wermer sendromudur. Bu sendromda paratiroid bez tümörleri (%90), pankreas adacık hücre tümörleri, ön hipofiz tümörleri birlikte görülebilmektedir. Bunun yanında adrenokortikal tü- mörler, karsinoidler, fasiyal anjiyofibromlar, kollajenomalar, lipomatöz tümörler, menenjiyomalar eşlik edebilmektedir. MEN Tip 2, diğer adı ile Sipple sendromu, önceleri MEN Tip 2A olarak tanımlanmaktaydı. Bu grupta medüller tiroid karsi- nomları, feokromasitoma ve paratiroid bez tümörleri olabilmektedir. MEN Tip 3, önceleri MEN Tip 2B olarak bilinirdi. Medüller tiroid karsinomu, feokromasitoma ve yanında marfanoid görünüm, mukozal nöromalar, intestinal otonomik gang- liyon disfonksiyonu eşlik edebilmektedir. Az oranda paratiroid bez tümörleri de görülebilir
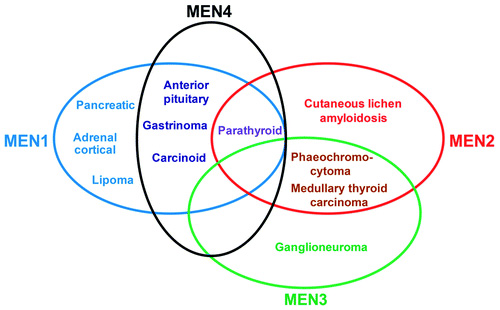
MEN1 hastalığı otozomal dominant kalıtılmakta olup, bir tümör süpresör gen olan MEN1 mutasyonu sonucu gelişir. MEN Tip 2-3’te otozomal dominant olarak kalıtılıp, RET proto-onkogen mutasyonu görülmektedir. Ratlarda saptanan MEN X hastalığında, hayvanların MEN 1-2 sendromlarının fenotiplerini gösterdiği sap Nöroendokrin tümörler (NET) diğer MEN sendromlarına göre daha düşük oranda MEN4 vakalarında görülebilmektedir. Bu vakalar genellikle gastrik-duo- denal ve pankreatik NET olup, fonksiyonel ya da non fonksiyonel olabilmektedir. Gastrinoma ve buna bağlı oluşan klinik sendrom Zollinger Ellison Sendromu 2 hastada tespit edilmiştir. VİPoma, glukagonoma gibi nöroendokrin tümörler ra- porlanmamıştır. MEN4 vakalarında rastlanan nöroendokrin tümörlerin MEN1 vakalarına göre çok daha düşük penetranslı olduğu bilinmektedir
Testis kanseri, nöroendokrin servikal karsinomlar, lipoma, anjiyofibroma, kol- lajenoma MEN1 hastalığı ile görülmesine karşın MEN4 vakalarında raporlanmamıştır.
Son yıllarda MEN sendromlarının tümör patogenezinin genetik ve moleküler mekanizmalarında ciddi bir ilerleme sağlanmıştır. Bu bölümde MEN ailesinin en yeni üyesi MEN4 sendromunun klinik ve genetik özelliklerinden bahsettik. MEN4 diğer MEN sendromlarının özelliklerini taşıyabilen, nadir görülen bir sendrom- dur. MEN4 oluşumuna neden olan CDKN1B mutasyonu keşfi ile birlikte tanısal anlamda önemli bir yol alınmıştır. MEN1 benzeri özelliklere ve negatif MEN1 tes- tine sahip vakalara, MEN4 için genetik danışmanlık ve test sunulmalıdır. Germ- line CDKN1B mutasyonunun tanımlanması, MEN4 için periyodik klinik, biyo- kimyasal ve radyolojik taramayı gerektirmektedir. CDKN1B somatik mutasyonları NET’lerde ve diğer endokrin olmayan neoplazilerde sıktır ve bu bilginin bu lez- yonlar için moleküler hedefli tedavilerde potansiyel kullanımına işaret etmektedir.
Multipl Endokrin Adenomatoz, Tip 4
Çoklu endokrin neoplazi, tip 4 (MEN 4), paratiroid bezlerinin adenomları ve bazen hiperplazisi ve pankreas adacık hücrelerinin ve/veya hipofiz bezinin tümörleri ile karakterize otozomal dominant bir sendromdur. Hiperparatiroidizm ve asemptomatik hiperkalsemi sıklıkla görülür. Tanı hormonal testler ve görüntüleme testleri ile konur. Semptomlara neden olan tümörler mümkün olduğunda cerrahi olarak çıkarılır. Tümörler semptomlara neden oluyorsa veya boyut kriterlerine göre malignite şüphesi varsa cerrahi olarak çıkarılır.
MEN 4’e , p27 veya p27KIP1 olarak da bilinen sikline bağımlı kinaz inhibitörü 1B proteinini kodlayan CDKN1B geninin etkisizleştirici bir mutasyonu neden olur . Protein hücresel büyüme ve gelişmede rol oynar ve hücre döngüsünün düzenlenmesinde önemli bir role sahiptir. Bir tümör baskılayıcı görevi görür ve protein kaybolduğunda hücreler planlanmamış çoğalmaya maruz kalabilir ve bu da sonunda kansere yol açabilir.
MEN 4, MEN 1 ile aynı birincil organları (paratiroidler, pankreas ve hipofiz) içermesine rağmen, hastalar MEN 1’li hastalara göre yaşamlarının ilerleyen dönemlerinde ortaya çıkma eğilimindedir ve hastalığın seyri daha yavaştır.
MEN 4, MEN 1’e göre çok daha az yaygındır ve şu anda dünya literatüründe tanımlanan hasta sayısı 100’den azdır.
MEN 4’ün klinik belirtileri , bazı farklılıklara rağmen esasen MEN 1’inkilerle aynıdır . MEN 4 ve hiperparatiroidizmli hastaların çoğunda tek paratiroid adenomları bulunurken, MEN 1’deki dört paratiroid bezinin tümünün hiperplazisi vardır. Hipofizde adrenokortikotropik hormon üreten adenomlar en sık görülen tümördür (%33), prolaktin salgılayan tümörler ise (%24) veya büyüme hormonu (%19) biraz daha az yaygındır. Pankreas nöroendokrin tümörleri gastrinoma (%33) veya işlevsiz tümörler (%66) olarak karakterize edilmiştir ( 1 ).
Serum kalsiyum, paratiroid hormonu (PTH), gastrin ve prolaktin düzeyleri
MEN 4 tanısı, MEN 1 ile aynı yaklaşımı izler ve hormon fazlalığı için kan testini ve CDKN1B genindeki nedensel mutasyonu belirlemek için genetik testi içerir . MEN 1 ve MEN 4 arasındaki klinik benzerlik nedeniyle, bu semptom grubuna sahip hastaların, hem MEN1 hem de CDKN1B’yi içeren bir gen paneli kullanılarak genetik teste tabi tutulması gerekir .
İlk laboratuvar çalışmaları serum kalsiyumu, paratiroid hormonu , gastrin ve prolaktin düzeylerini içerir. Bu testler MEN 1’e bağlı bir endokrin anormalliği düşündürürse ek laboratuvar veya radyolojik testlere ihtiyaç duyulabilir.
Pankreas veya duodenumun gastrin salgılayan gastroenteropankreatik nöroendokrin (GEP-NET) tümörü, yüksek bazal plazma gastrin seviyeleri, infüze edilen kalsiyuma abartılı bir gastrin tepkisi ve sekretin infüzyonundan sonra gastrin seviyesinde paradoksal bir artış ile teşhis edilir . Pankreasın insülin salgılayan beta hücreli tümörü , açlık hipoglisemisinin yüksek plazma insülin seviyesiyle tespit edilmesiyle teşhis edilir. Pankreatik polipeptit veya gastrinin yüksek bazal seviyesi veya bu hormonların standart bir öğüne aşırı tepkisi, pankreas tutulumunun en erken belirtisi olabilir.
Akromegali, glikoz uygulamasıyla baskılanamayan yüksek büyüme hormonu seviyeleri ve serum insülin benzeri büyüme faktörü 1’in (somatomedin C) yüksek seviyeleri ile teşhis edilir.
Ultrasonografi veya bilgisayarlı tomografi (BT) tümörlerin lokalizasyonuna yardımcı olabilir. Bu tümörler genellikle küçük olduğundan ve lokalizasyonu zor olduğundan, diğer görüntüleme testleri (örneğin, sarmal [spiral] BT, anjiyografi, MR, endoskopik ultrasonografi, intraoperatif ultrasonografi) gerekli olabilir. Flor-18 [18F] etiketli deoksiglukoz (18F-FDG) veya galyum Ga 68 dotatat pozitron emisyon tomografisi (PET)/BT ile torasik görüntüleme, bronkopulmoner nöroendokrin tümörleri benign pulmoner nodüllerden ayırmada ve timik karsinoidin tanımlanmasında yararlı olabilir. Pankreatik ve duodenal nöroendokrin tümörler için, galyum Ga 68 dotatat pozitron emisyon tomografisi/BT , 26 MEN 1 vakasında çoklu görüntüleme modalitelerinin yer aldığı bir çalışmada oktreotid taramasından veya CT taramasından üç kat daha duyarlıydı ; Mümkün olduğunda bu test, periyodik görüntüleme için oktreotid taramasının yerini almalıdır .
MEN 4’ün tedavisi MEN 1’in tedavisine benzer ve mümkün olduğunda tümör rezeksiyonuyla başlar. MEN 4 ve hiperparatiroidizmli hastalarda tipik olarak tek bir adenom sergilendiğinden, ameliyattan önce tek bir adenom tanımlanırsa diğer bezlerin araştırılması ve/veya çıkarılması gerekli olmayabilir.
Tümörlerin tamamen çıkarılması mümkün olmadığında hormon fazlalığını tedavi etmek için ilaçlar kullanılır. Bunlar şunları içerir:
Hiperparatiroidizme bağlı tekrarlayan veya kalıcı hiperkalsemi tedavisinde Octreotide ve sinakalset
Prolaktin salgılayan hipofiz tümörlerine bağlı hiperprolaktinemi tedavisinde dopamin agonistleri
Hipergastrinemiye bağlı semptomatik peptik ülser hastalığının semptomatik kontrolü için proton pompa inhibitörleri
İnsülinoma bağlı hipoglisemi için diazoksit veya somatostatin analoğu ( oktreotid , lanreotid )
Tümör yükünü azaltmak için Streptozosin veya diğer sitotoksik ilaçlar kullanılabilir.
Somatostatin analogları aynı zamanda karsinoid tümörler dahil gastrin salgılamayan diğer pankreas tümörlerinden hormon salgılanmasını da bloke edebilir ve iyi tolere edilir.
Metastatik pankreas tümörleri için palyatif tedaviler arasında hepatik kitle küçültme ameliyatı ve hepatik arter kemoembolizasyonu yer alır.