Gastrointestinal Polipozis Sendromlarının hangilerinde intestinal hamartomatöz polip görülür?
A) Peutz-Jeghers sendromu**********
B) Gardner sendromu
C) Turcot sendromu
D) Ailesel adenomatoz polipozis (FAP) sendromu
E) MUTYH İlişkili Polipozis (MAP) sendromu
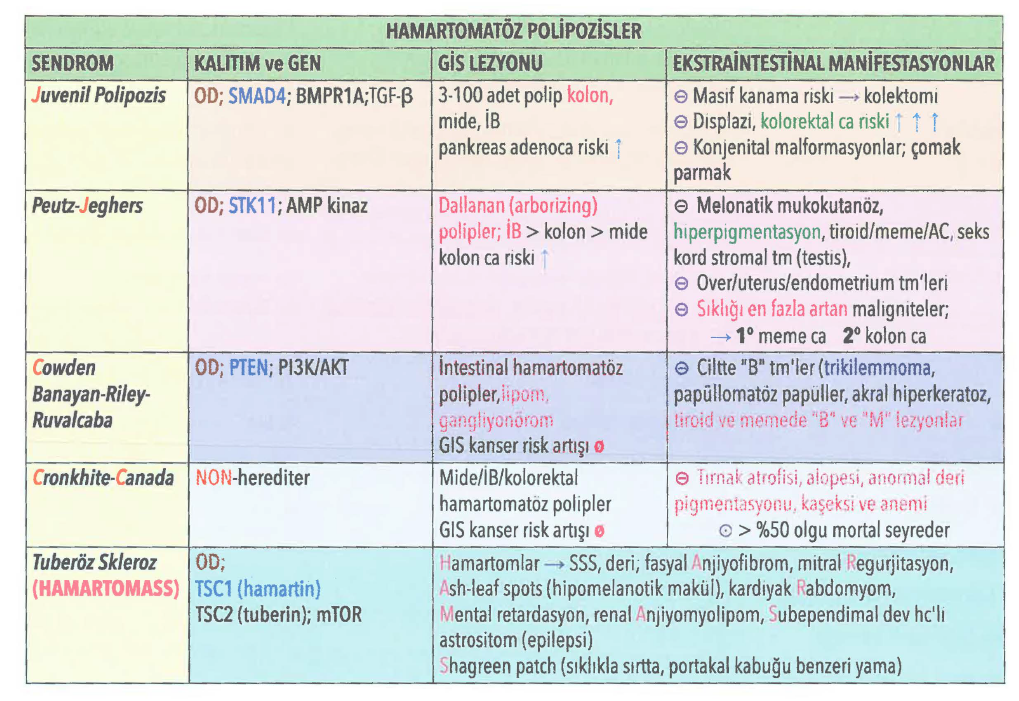
HAMARTOMATÖZ POLİP SENDROMLARI
1-Juvenil polipozis sendromu, “Retansiyon polipleri”; Multipl jüvenil polip riskini artırır, kolon adenokarsinomu ve GİS’te pankreas kanseri, SMAD4 ve BMPR1A mutasyonları görülebilir.
2-Peutz Jeghers sendromu, PJS; GİS’te multipl hamartomatöz polipler, mukokutanöz pigmentasyon, STK 11 mutasyonu yaygın; GIS, testis, yumurtalık, rahim, pankreas, meme kanseri riski.
3-“Multipl Hamartom Sendromu”, Cowden sendromu; PTEN gen mutasyonu var, OD geçiyor. Mide, ince barsak ve kolonda hamartomatöz polipler, yemek borusunda glukojenik akantoz, trichilemmoma, akral keratoz, oral papillomlar, memede fibroadenom ve fibrokistik değişiklikler, tiroidde guatr ve adenomaglimalar, makrosenfasit görülebilir. GİS’te malignite riski artmaz, ancak meme ve tiroid kanseri riski artar.
4-Bannayan-Riley-Ruvalcaba sendromu, BRRS; PTEN gen mutasyonuna bağlı gelişir. Makrosefali multipl hemanjiyomlar, lipomatozis, genital pigmentasyonlar, lipid miyopati ve intestinal hamartomatöz polipler vardır.
5-Gastrointestinal polipozis-cilt pigmentasyonu-alopesi-tırnak değişiklikleri sendromu “ Cronkhite-Canada sendromu; Nedeni bilinmeyen edinilmiş bir hastalıktır. 50 yaşından sonra görülür. GİS’te hamartomatöz polipler, tırnak distrofisi, alopesi ve deri hiperpigmentasyonu görülür. Hastalarda protein kaybettiren enteropati, anemi, elektrolit bozuklukları olabilir. Hastaların çoğu kaşeksiden muzdariptir. kayıp.
6-“Tuberous skleroz kompleksi (TSC), Bourneville hastalığı”, Tuberoskleroz TSC1 (hamartin) ve TSC2 (tuberin) gen mutasyonlarına bağlı olarak gelişir. GİS’te hamartomatöz polipler, zeka geriliği, epilepsi, renal anjiyomiyolipom, fasiyal anjiyofibrom görülebilir.
ADENOMATÖZ POLİP SENDROMLARI
1-“Ailevi adenomatöz polipozis”, FAP sendromu; APC mutasyonu, kolon, ince barsak ve midede adenomatöz polipler, midede funfik bez palipleri, %100 kolon adenokarsinomu gelişme riski
2- “Ailesel kolorektal polipozis, Kolonun ailesel polipozisi” Gardner sendromu; APC mutasyonu, kolon adenomları, kafatası ve çene osteomları, dermoid kist, desmoid tümörler
3-Turcot sendromu, Mismatch onarım kanseri sendromu, Beyin tümörü-polipoz sendromu, Glioma-polipoz sendromu; APC mutasyonu, kolon adenomları, glioblastoma, medulloblastoma
4-MAP sendromu; MUTYH “mutY DNA glikosilaz” mutasyonu, kolonda Adenomatöz Polipler, sapsız tırtıklı adenomlar, hiperplastik polipler
5- “Kalıtsal nonpolipozis kolorektal kanser” HNPCC (Lynch sendromu) MLH1, MSH2 mutasyonu, sağ kolon ve çekumda adenokarsinom, endometriyum ve yumurtalık kanserleri
Hamartamatöz polipler içinde kolon kanseri yapmayan hangisidir?
A) Juvenil polip
B)Peutz-Jegers sendromu
C) Cowden sendromu
D) Bannayan-Ruvalcaba-Riley sendromu
E) Cronkhite Canada sendromu
Cevap: E
Hamartamatöz polipler içinde sadece 4 tanesi kolon ca yapıyor. (A,B,C,D şıkları)
Cronkhite canada sendromunda kolon ca gelişmez.
Hamartomatöz Polipler
Jüvenil polipler: Sıklıkla beş yaşından önce izlenirler. Lamina proprianın hamartomatöz proliferasyonuyla karekterizedir. İleri yaşlarda görüldüklerinde ‘retansiyon polibi’ olarak adlandırılırlar. Yetişkinlerde görülenler inflamatuar poliple ayrılmazlar. Sıklıkla rektumda yerleşir, 3 cmden küçüktür ve tektir. Malign potansiyeli yoktur. Kanama ve bazı olgularda ağrılı infarktüsleri izlenir. Otoampütasyon izlenebilir. OD geçişli juvenil polipozis sendromunda multiple polipler izlenir ve aşırı kanama nedeniyle kolektemi uygulanabilir. Bu sendromlu hastalarda az da olsa kolon kanseri riski vardır.
Peutz-Jeghers polipleri: Sık olmayan, hamartomatöz poliplerdir. Malignite potansiyeli yoktur. Ancak otozomal
dominant geçişli Peutz – Jeghers sendromunda melanotik mukozal ve kutanöz pigmentasyon ile beraber bulunur ve intestinal ve ekstraintestinal malignite gelişme riski anlamlı oranda yüksektir. Bu sendromda polipler sıklıkla ince barsakta izlenir. İleum, jejunum ve midede hamartomatöz polipler ve ek organlarda kanserlerle karekterize bir hastalıktır. 19. kromozomda bulunan serin threonin kinaz aktivitesi gösteren gende (STK11) mutasyon vardır. Deri ve mukozlarda hiperpigmentasyon izlenir. Kanser gelişme riski artan organlar: deri, pankreas, meme, akciğer, over ve uterus kanserleridir. Pankreas kanseri riskini 120 kat artırır!
Bu poliplerde lamina propriada düz kas hücrelerinin bulunması tipiktir. Juvenil poliplerden ayırımında en önemli farkı budur.
Cronkhite-Canada Sendromu polipleri: Herediter değildir. 50 yaşından sonra izlenir. Barsaklarda hamartomatöz polipler ve bunlara eşlik eden ektodermal anomalilerle karekterizedir. Eşlik eden ektodermal anomaliler hiperpigmentasyon, vitiligo, alopesi, tırnaklarda atrofik değişikliklerdir. Juvenil poliplerden histolojik olarak ayrılamayan multiple polipler vardır. Fakata hastaların % 50 si kaşeksi ve anemiden ölür.
Cowden Sendromu polipleri: Otozomal dominant geçişlidir. Multiple hamartomatöz polip, fasiyal trikilemmoma, akral keratoz, oral papillomlarla, glans peniste makül,hemanjiyomlar ve makrosefali ile karekterizedir. Bu sendromda tumor supresor gen olan PTEN geninde kalıtsal mutasyon vardır. GIS malignite riski artışı izlenmez fakat meme, foliküler tiroid ve endometriyal kanser riski artışı izlenir. Bannayan-Ruvalcaba-Riley sendromu; Cowden sendromuyla aynıdır fakat farkları mental ve büyüme geriliğinin olması, neoplazi riskinin daha az artmış olmasıdır.
Tuberoskleroz sendromu polipleri: Hamartomatöz rektal poliplerle karekterizedir.
PTEN gen mutasyonu: Cowden sendromu
Kalıtsal olmayan polipozis sendromu: Cronkhite-Canada sendromu
STK, LKB mutasyonu: Peutz Jeghers sendromu
En sık izlenen polip: Hiperplastik polip
Hamartomatöz poliplerin izlendiği sendromlar: Juvenil Polipozis sendromu, Peutz Jeghers sendromu, Cowden
sendromu, Cronkhite Canada sendromu, Tuberoskleroz sendromu